Release of Pharmaceutical Peptides in an Aggregated State: Using Fibrillar Polymorphism to Modulate Release Levels


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. The Peptide 3474 Aggregates in Buffer and in the Presence of Different Surfactants
3.2. 3474 Aggregates Show Burst Release of Soluble Peptides, Particularly Those Formed in DDM
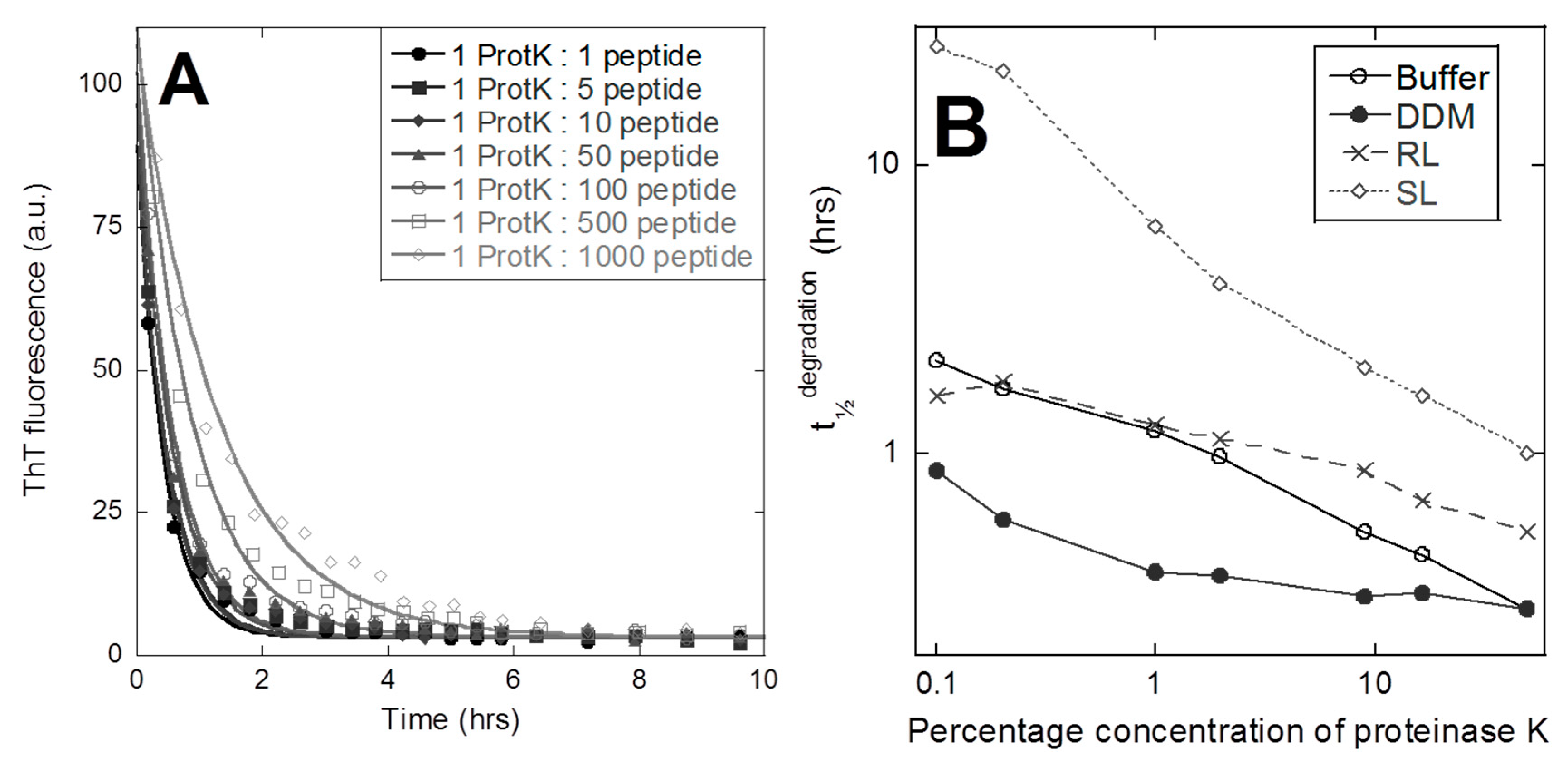
3.3. Fibril Structure Differences is Reflected by Significant Differences in Proteolysis Susceptibility
4. Discussion and Conclusions
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
Abbreviations
| SDS | sodium dodecyl sulphate |
| DDM | dodecyl maltoside |
| RL | rhamnolipid |
| SL | sophorolipid |
| NaP | sodium phosphate |
| PBS | phosphate buffered saline |
| 3474 | glucagon analogue |
References
- Goosen, M.F.; Leung, Y.F.; Chou, S.; Sun, A.M. Insulin-albumin microbeads: An implantable, biodegradable system. Biomater. Med. Devices Artif. Organs 1982, 10, 205–218. [Google Scholar] [CrossRef]
- Garbayo, E.; Ansorena, E.; Lanciego, J.L.; Aymerich, M.S.; Blanco-Prieto, M.J. Sustained release of bioactive glycosylated glial cell-line derived neurotrophic factor from biodegradable polymeric microspheres. Eur. J. Pharm. Biopharm. 2008, 69, 844–851. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Dong, Q.; Wang, M.; Shi, L.; Wu, Y.; Yu, X.; Shi, Y.; Shan, Y.; Jiang, C.; Zhang, X.; et al. Preparation, characterization, and pharmacodynamics of exenatide-loaded poly(DL-lactic-co-glycolic acid) microspheres. Chem. Pharm. Bull. (Tokyo) 2010, 58, 1474–1479. [Google Scholar] [CrossRef]
- Kamath, K.R.; Park, K. Biodegradable hydrogels in drug delivery. Adv. Drug Deliv. Rev. 1993, 11, 59–84. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef] [Green Version]
- Haghjou, N.; Soheilian, M.; Abdekhodaie, M.J. Sustained release intraocular drug delivery devices for treatment of uveitis. J. Ophthalmic. Vis. Res. 2011, 6, 317–329. [Google Scholar] [PubMed]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef]
- Freiberg, S.; Zhu, X.X. Polymer microspheres for controlled drug release. Int. J. Pharm. 2004, 282, 1–18. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, H.; He, W.; Zhao, D.; Song, A.; Luan, Y. Disulfide-Linked Amphiphilic Polymer-Docetaxel Conjugates Assembled Redox-Sensitive Micelles for Efficient Antitumor Drug Delivery. Biomacromolecules 2016, 17, 1621–1632. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, P.; Zhao, Q.; Zhang, Y.; Cao, L.; Luan, Y. Doxorubicin-loaded polypeptide nanorods based on electrostatic interactions for cancer therapy. J. Colloid Interface Sci. 2016, 464, 126–136. [Google Scholar] [CrossRef]
- Rogovina, S.Z. Biodegradable polymer composites based on synthetic and natural polymers of various classes. Prog. Polym. Sci. 2016, 58, 1–26. [Google Scholar] [CrossRef]
- Vora, J.; Cariou, B.; Evans, M.; Gross, J.L.; Harris, S.; Landstedt-Hallin, L.; Mithal, A.; Rodriguez, M.R.; Meneghini, L. Clinical use of insulin degludec. Diabetes Res. Clin. Pract. 2015, 109, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Seuring, C.; Nespovitaya, N.; Rutishauser, J.; Spies, M.; Riek, R. Hormone amyloids in sickness and in health. In Amyloid Fibrils and Prefibrillar Aggregates; Otzen, D.E., Ed.; WileyVCH Verlag & Co. KGaA: Berlin/Heidelberg, Germany, 2013; pp. 395–410. [Google Scholar]
- Otzen, D.E.; Nielsen, P.H. We find them here, we find them there: Functional bacterial amyloid. Cell. Mol. Life Sci. 2008, 65, 910–927. [Google Scholar] [CrossRef] [PubMed]
- Christensen, L.F.B.; Schafer, N.; Wolf-Perez, A.; Madsen, D.J.; Otzen, D.E. Bacterial amyloids: Biogenesis and biomaterials. In Biological and Bio-Inspired Nanomaterials: Assembly Mechanisms and Properties; Perrett, S., Buell, A.K., Knowles, T.J., Eds.; Springer Science and Business Media: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- LeVine, H., 3rd. Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999, 309, 274–284. [Google Scholar]
- O’Nuallain, B.; Shivaprasad, S.; Kheterpal, I.; Wetzel, R. Thermodynamics of Aβ(1−40) Amyloid Fibril Elongation. Biochemistry 2005, 44, 12709–12718. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Costa, P.R.; Griffin, R.G.; Lansbury, P.T. Models of the beta Protein C-Terminus: Differences in Amyloid Structure May Lead to Segregation of “Long” and “Short” Fibrils. J. Am. Chem. Soc. 1994, 116, 9741–9742. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Knowles, T.P.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Waudby, C.A.; Mossuto, M.F.; Meehan, S.; Gras, S.L.; et al. Metastability of native proteins and the phenomenon of amyloid formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef]
- Gazit, E. The “Correctly Folded” state of proteins: Is it a metastable state? Angew. Chem. Int. Ed. Engl. 2002, 41, 257–259. [Google Scholar] [CrossRef]
- Dobson, C.M. The structural basis of protein folding and its links with human disease. Philos. Trans. R. Soc. Lond B Biol. Sci. 2001, 356, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Morel, B.; Varela, L.; Conejero-Lara, F. The thermodynamic stability of amyloid fibrils studied by differential scanning calorimetry. J. Phys. Chem. B 2010, 114, 4010–4019. [Google Scholar] [CrossRef]
- Adler-Abramovich, L.; Reches, M.; Sedman, V.L.; Allen, S.; Tendler, S.J.; Gazit, E. Thermal and chemical stability of diphenylalanine peptide nanotubes: Implications for nanotechnological applications. Langmuir 2006, 22, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Fitzpatrick, A.W.; Meehan, S.; Mott, H.R.; Vendruscolo, M.; Dobson, C.M.; Welland, M.E. Role of intermolecular forces in defining material properties of protein nanofibrils. Science 2007, 318, 1900–1903. [Google Scholar] [CrossRef] [PubMed]
- Nordstedt, C.; Naslund, J.; Tjernberg, L.O.; Karlstrom, A.R.; Thyberg, J.; Terenius, L. The Alzheimer A beta peptide develops protease resistance in association with its polymerization into fibrils. J. Biol. Chem. 1994, 269, 30773–30776. [Google Scholar] [PubMed]
- Maji, S.K.; Schubert, D.; Rivier, C.; Lee, S.; Rivier, J.E.; Riek, R. Amyloid as a depot for the formulation of long-acting drugs. PLoS Biol. 2008, 6, e17. [Google Scholar] [CrossRef]
- Otzen, D.E. Amyloid formation in surfactants and alcohols: Membrane mimetics or structural switchers? Curr. Protein Pept. Sci. 2010, 11, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Giehm, L.; Oliveira, C.L.; Christiansen, G.; Pedersen, J.S.; Otzen, D.E. SDS-induced fibrillation of alpha-synuclein: An alternative fibrillation pathway. J. Mol. Biol. 2010, 401, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Leffers, K.W.; Schell, J.; Jansen, K.; Lucassen, R.; Kaimann, T.; Nagel-Steger, L.; Tatzelt, J.; Riesner, D. The Structural Transition of the Prion Protein into its Pathogenic Conformation is Induced by Unmasking Hydrophobic Sites. J. Mol. Biol. 2004, 344, 839–853. [Google Scholar] [CrossRef]
- Andersen, K.K.; Oliveira, C.L.P.; Larsen, K.L.; Poulsen, F.M.; Callisen, T.H.; Westh, P.; Pedersen, J.S.; Otzen, D.E. The role of decorated SDS micelles in sub-cmc protein denaturation and association. J. Mol. Biol. 2009, 391, 207–226. [Google Scholar] [CrossRef]
- Patil, S.M.; Xu, S.; Sheftic, S.R.; Alexandrescu, A.T. Dynamic alpha-helix structure of micelle-bound human amylin. J. Biol. Chem. 2009, 284, 11982–11991. [Google Scholar] [CrossRef]
- Knight, J.D.; Miranker, A.D. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 2004, 341, 1175–1187. [Google Scholar] [CrossRef]
- Chirita, C.N.; Necula, M.; Kuret, J. Anionic micelles and vesicles induce tau fibrillization in vitro. J. Biol. Chem. 2003, 278, 25644–25650. [Google Scholar] [CrossRef]
- Loureiro, J.A.; Rocha, S.; Mdo, C.P. Charged surfactants induce a non-fibrillar aggregation pathway of amyloid-beta peptide. J. Pept. Sci. 2013, 19, 581–587. [Google Scholar] [CrossRef]
- Lougheed, W.D.; Albisser, A.M.; Martindale, H.M.; Chow, J.C.; Clement, J.R. Physical stability of insulin formulations. Diabetes 1983, 32, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Bam, N.B.; Cleland, J.L.; Yang, J.; Manning, M.C.; Carpenter, J.F.; Kelley, R.F.; Randolph, T.W. Tween protects recombinant human growth hormone against agitation-induced damage via hydrophobic interactions. J. Pharm. Sci. 1998, 87, 1554–1559. [Google Scholar] [CrossRef] [PubMed]
- Bam, N.B.; Cleland, J.L.; Randolph, T.W. Molten globule intermediate of recombinant human growth hormone: Stabilization with surfactants. Biotechnol. Prog. 1996, 12, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, N.; Nielsen, S.B.; Buell, A.K.; Kaspersen, J.D.; Arosio, P.; Vad, B.S.; Paslawski, W.; Christiansen, G.; Valnickova-Hansen, Z.; Andreasen, M.; et al. The role of stable α-synuclein oligomers in the molecular events underlying amyloid formation. J. Am. Chem. Soc. 2014, 136, 3859–3868. [Google Scholar] [CrossRef]
- Zandomeneghi, G.; Krebs, M.R.; McCammon, M.G.; Fandrich, M. FTIR reveals structural differences between native beta-sheet proteins and amyloid fibrils. Protein Sci. 2004, 13, 3314–3321. [Google Scholar] [CrossRef]
- Steiner, S.S.; Li, M.; Hauser, R.; Pohl, R. Stabilized glucagon formulation for bihormonal pump use. J. Diabetes Sci. Technol. 2010, 4, 1332–1337. [Google Scholar] [CrossRef]
- Madsen, J.K.; Pihl, R.; Møller, A.L.B.; Madsen, A.T.; Otzen, D.E.; Andersen, K.K. The anionic biosurfactant rhamnolipid does not denature industrial enzymes. Front. Microbiol. 2015, 6, 292. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.K.; Otzen, D.E. Denaturation of a-lactalbumin and myoglobin by the anionic biosurfactant Rhamnolipid. Biochim. Biophys. Acta 2014, 1844, 2338–2345. [Google Scholar] [CrossRef] [PubMed]
- Madsen, J.K.; Giehm, L.; Otzen, D.E. The use of surfactants to solubilise a glucagon analogue. Pharm. Res. 2018, 35, 235. [Google Scholar] [CrossRef]
- Malmos, K.G.; Bjerring, M.; Jessen, C.M.; Nielsen, E.H.; Poulsen, E.T.; Christiansen, G.; Vosegaard, T.; Skrydstrup, T.; Enghild, J.J.; Pedersen, J.S.; et al. How Glycosaminoglycans Promote Fibrillation of Salmon Calcitonin. J. Biol. Chem. 2016, 291, 16849–16862. [Google Scholar] [CrossRef] [PubMed]
- Janciauskiene, S.; de Frutos, P.G.; Carlemalm, E.; Dahlback, B.; Eriksson, S. Inhibition of Alzheimer beta-peptide fibril formation by serum amyloid P component. J. Biol. Chem. 1995, 270, 26041–26044. [Google Scholar] [CrossRef] [PubMed]
- Fradkin, A.H.; Carpenter, J.F.; Randolph, T.W. Immunogenicity of aggregates of recombinant human growth hormone in mouse models. J. Pharm. Sci. 2009, 98, 3247–3264. [Google Scholar] [CrossRef]
- Weltzien, R.B.; Pachter, J.S. Visualization of beta-amyloid peptide (Abeta) phagocytosis by human mononuclear phagocytes: Dependency on Abeta aggregate size. J. Neurosci. Res. 2000, 59, 522–527. [Google Scholar] [CrossRef]
- Gamble, C.N. The role of soluble aggregates in the primary immune response of mice to human gamma globulin. Int. Arch. Allergy Appl. Immunol. 1966, 30, 446–455. [Google Scholar] [CrossRef]
- van Beers, M.M.; Sauerborn, M.; Gilli, F.; Brinks, V.; Schellekens, H.; Jiskoot, W. Oxidized and aggregated recombinant human interferon beta is immunogenic in human interferon beta transgenic mice. Pharm. Res. 2011, 28, 2393–2402. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, A.J.; Krc, J., Jr.; Kinkel, A.W.; Samyn, J.C. Effect of polymorphism on the absorption of chloramphenicol from chloramphenicol palmitate. J. Pharm. Sci. 1967, 56, 847–853. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Dikov, D.; Flink, J.L.; Hjuler, H.A.; Christiansen, G.; Otzen, D.E. The changing face of glucagon fibrillation: Structural polymorphism and conformational imprinting. J. Mol. Biol. 2006, 355, 501–523. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Andersen, C.B.; Otzen, D.E. Amyloid structure—One but not the same: The many levels of fibrillar polymorphism. FEBS J. 2010, 277, 4591–4601. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Otzen, D.E. Amyloid—A state in many guises: Survival of the fittest fibril fold. Protein Sci. 2008, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R. Amyloid Polymorphism: Structural Basis and Neurobiological Relevance. Neuron 2015, 86, 632–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, J.S. The nature of amyloid-like glucagon fibrils. J. Diabetes Sci. Technol. 2010, 4, 1357–1367. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madsen, J.K.; Christiansen, G.; Giehm, L.; Otzen, D.E. Release of Pharmaceutical Peptides in an Aggregated State: Using Fibrillar Polymorphism to Modulate Release Levels. Colloids Interfaces 2019, 3, 42. https://doi.org/10.3390/colloids3010042
Madsen JK, Christiansen G, Giehm L, Otzen DE. Release of Pharmaceutical Peptides in an Aggregated State: Using Fibrillar Polymorphism to Modulate Release Levels. Colloids and Interfaces. 2019; 3(1):42. https://doi.org/10.3390/colloids3010042
Chicago/Turabian StyleMadsen, Jens K., Gunna Christiansen, Lise Giehm, and Daniel E. Otzen. 2019. "Release of Pharmaceutical Peptides in an Aggregated State: Using Fibrillar Polymorphism to Modulate Release Levels" Colloids and Interfaces 3, no. 1: 42. https://doi.org/10.3390/colloids3010042