Geno-Spatial Distribution of Mycobacterium Tuberculosis and Drug Resistance Profiles in Myanmar–Thai Border Area

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Setting
2.2. Study Design and Participants
2.3. Samples Collection, Genomic DNA Extraction, and Whole Genome Sequencing
2.4. Genome Sequencing Data and Variant Calling
2.5. Phylogenetic Analysis
2.6. Genotyping
2.7. Prediction of Drug Resistance
2.8. Statistical Analysis
2.9. Ethics
3. Results
3.1. Socio-Demographic Information
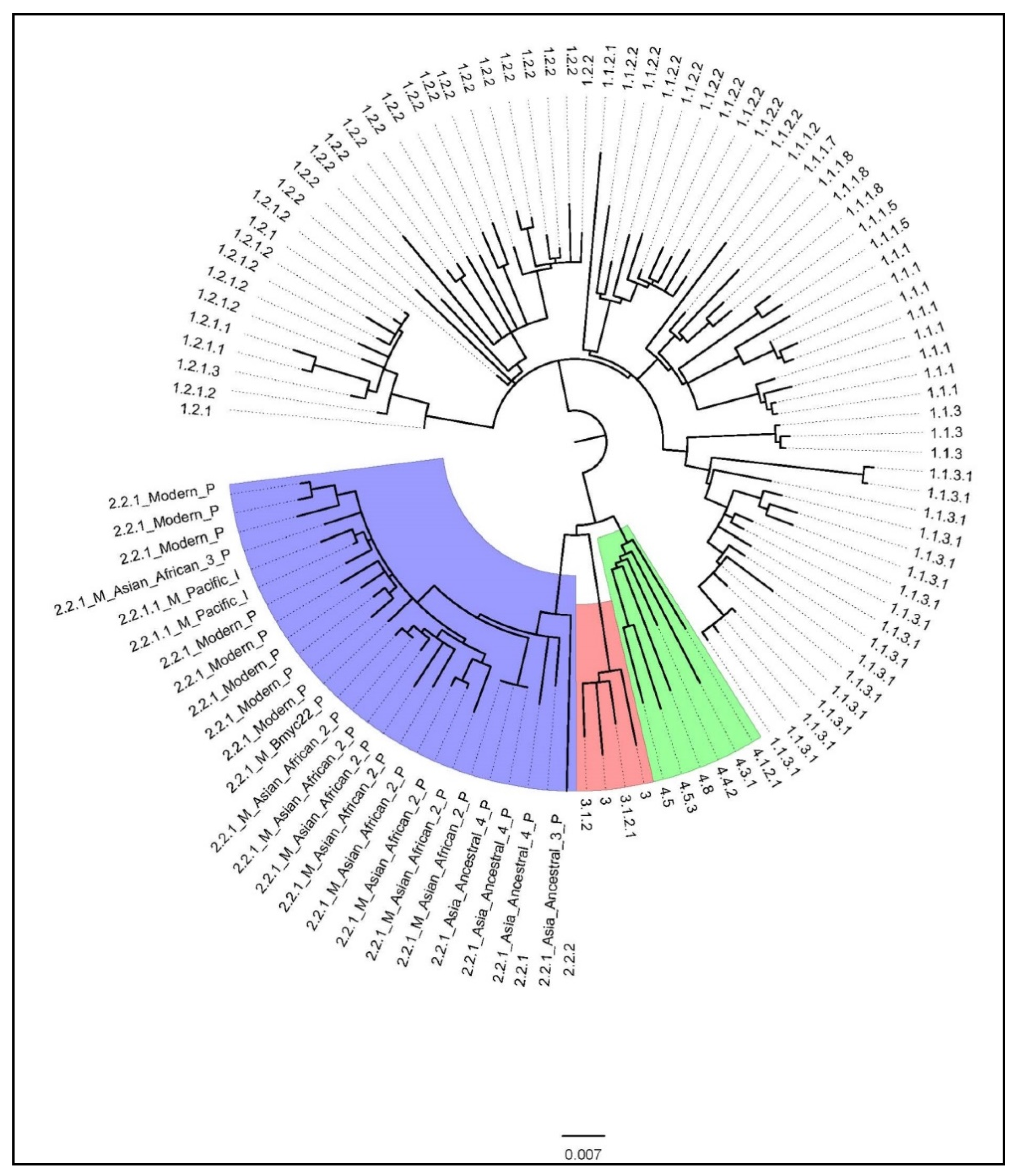
3.2. Genotype Information
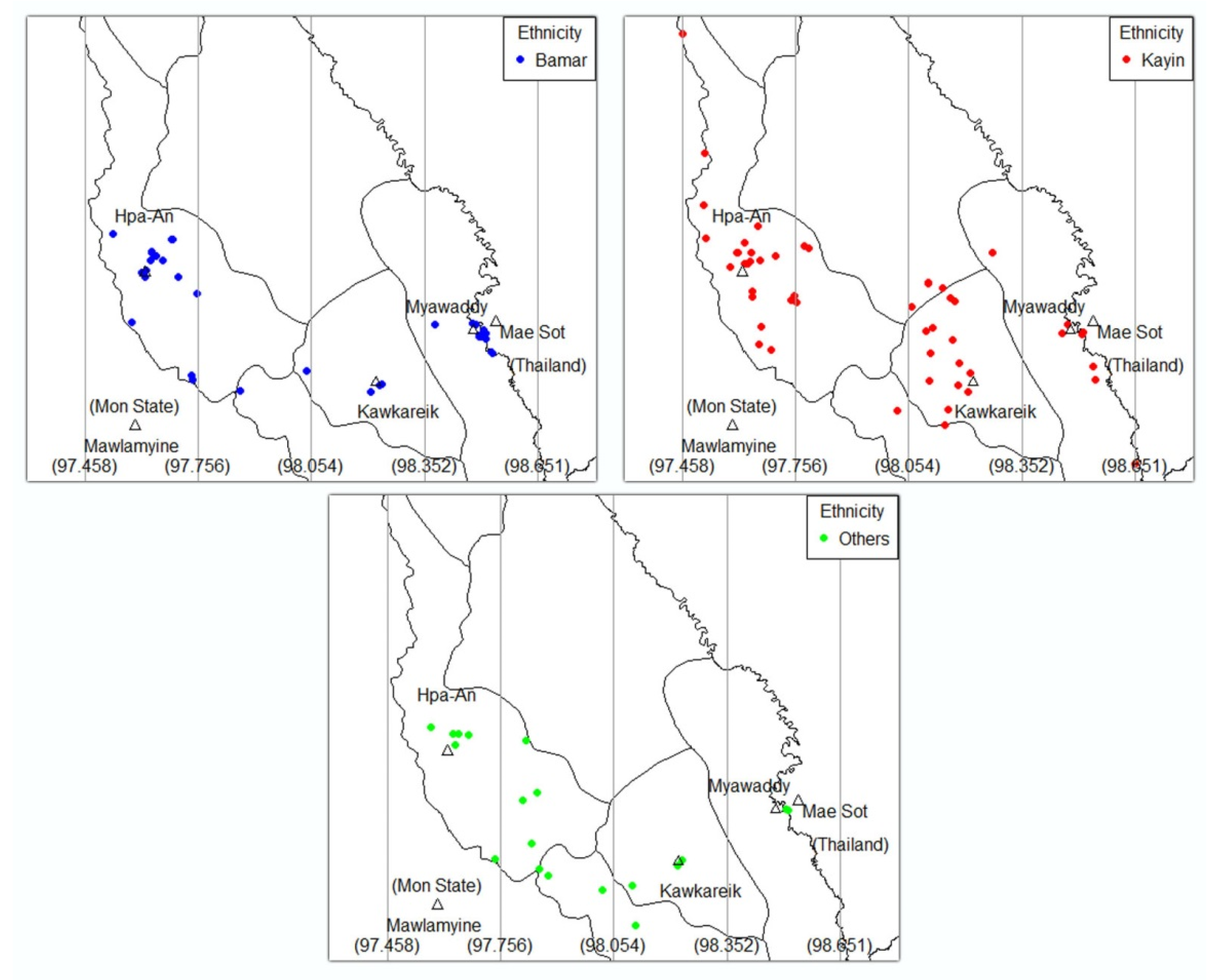
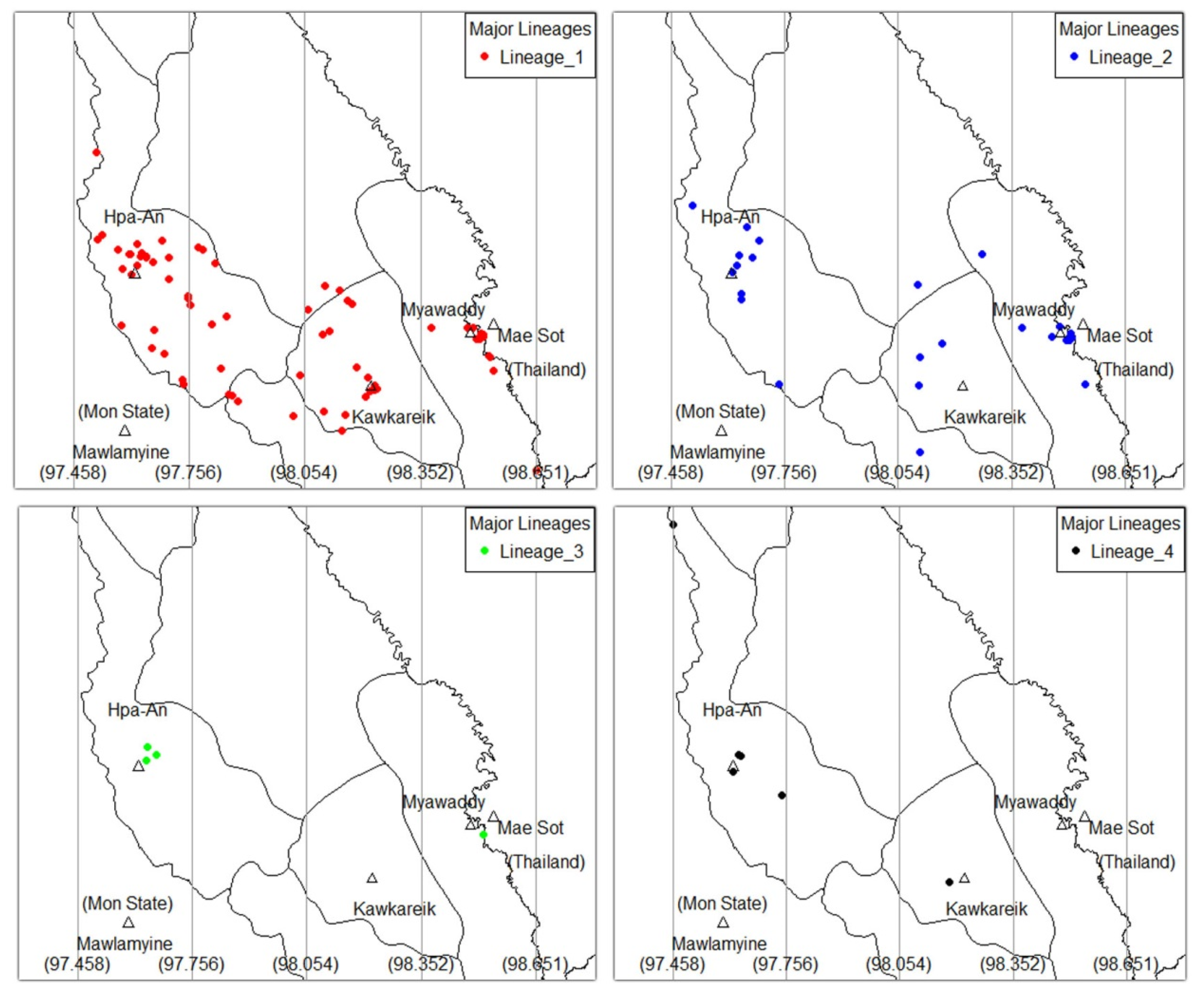
3.3. Geno-Spatial Analysis
3.4. Clinical Characteristics
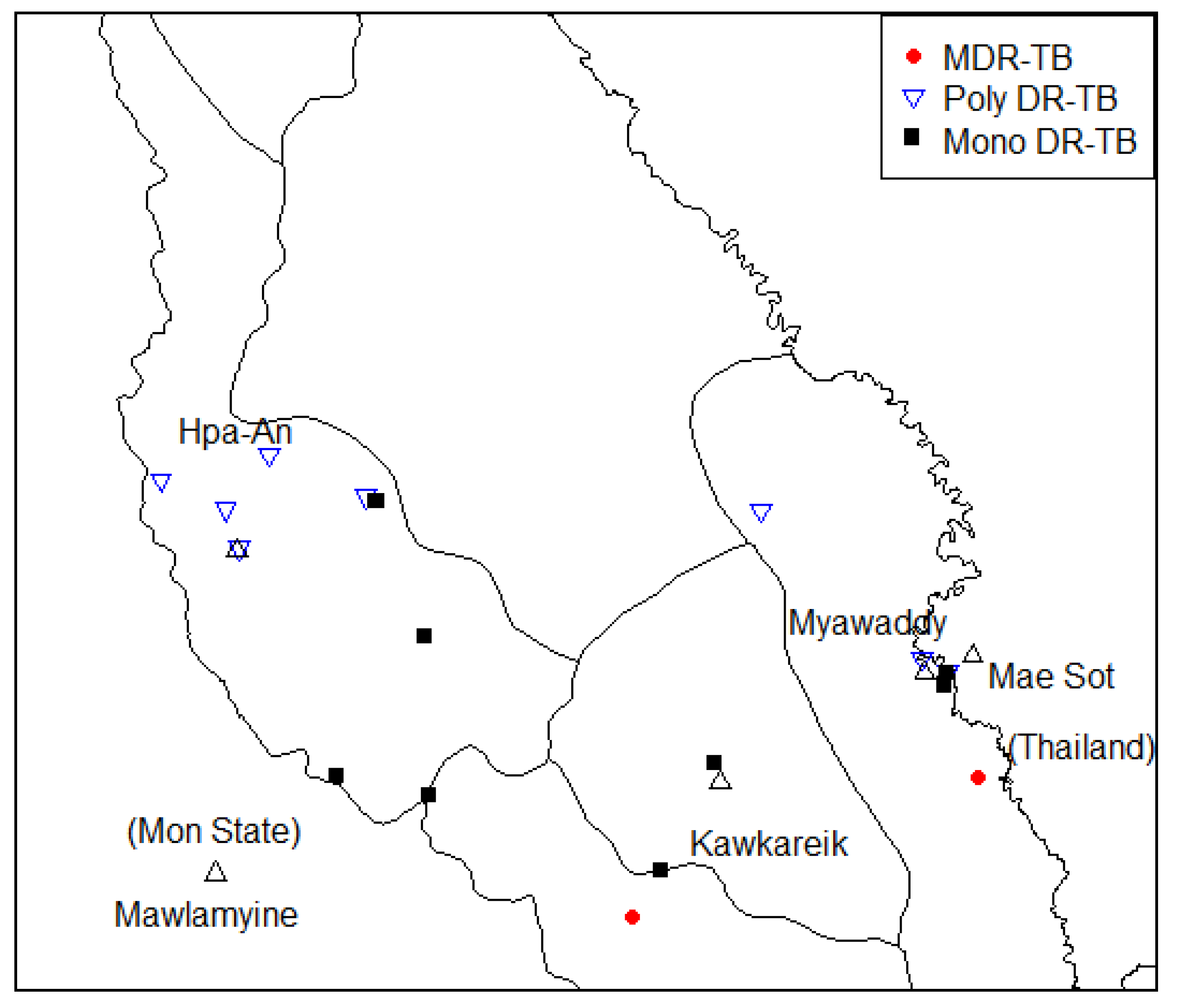
3.5. Drug Resistance of TB Drug Information
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2019. Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 23 April 2020).
- National Tuberculosis Programme. Annual TB Report, 2017; National Tuberculosis Programme: Yangon, Myanmar, 2018.
- Regmi, S.M.; Chaiprasert, A.; Kulawonganunchai, S.; Tongsima, S.; Coker, O.O.; Prammananan, T.; Viratyosin, W.; Thaipisuttikul, I. Whole genome sequence analysis of multidrug-resistant Mycobacterium tuberculosis Beijing isolates from an outbreak in Thailand. Mol. Genet. Genom. 2015, 290, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Schürch, A.C.; Kremer, K.; Daviena, O.; Kiers, A.; Boeree, M.J.; Siezen, R.J.; van Soolingen, D. High-Resolution Typing by Integration of Genome Sequencing Data in a Large Tuberculosis Cluster. J. Clin. Microbiol. 2010, 48, 3403–3406. [Google Scholar] [CrossRef] [Green Version]
- Cowan, L.S.; Mosher, L.; Diem, L.; Massey, J.P.; Crawford, J.T. Variable-Number Tandem Repeat Typing of Mycobacterium tuberculosis Isolates with Low Copy Numbers of IS6110 by Using Mycobacterial Interspersed Repetitive Units. J. Clin. Microbiol. 2002, 40, 1592–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kardan-Yamchi, J.; Kazemian, H.; Battaglia, S.; Abtahi, H.; Foroushani, A.R.; Hamzelou, G.; Cirillo, D.M.; Ghodousi, A.; Feizabadi, M.M. Whole Genome Sequencing Results Associated with Minimum Inhibitory Concentrations of 14 Anti-Tuberculosis Drugs among Rifampicin-Resistant Isolates of Mycobacterium Tuberculosis from Iran. J. Clin. Med. 2020, 9, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phyu, S.; Stavrum, R.; Lwin, T.; Svendsen, Ø.S.; Ti, T.; Grewal, H.M. Predominance of Mycobacterium tuberculosis EAI and Beijing Lineages in Yangon, Myanmar. J. Clin. Microbiol. 2009, 47, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbugi, E.V.; Katale, B.Z.; Streicher, E.M.; Keyyu, J.D.; Kendall, S.L.; Dockrell, H.M.; Michel, A.L.; Rweyemamu, M.M.; Warren, R.M.; Matee, M.I.; et al. Mapping of Mycobacterium tuberculosis Complex Genetic Diversity Profiles in Tanzania and Other African Countries. PLoS ONE 2016, 11, e0154571. [Google Scholar] [CrossRef] [Green Version]
- Ajawatanawong, P.; Yanai, H.; Smittipat, N.; Disratthakit, A.; Yamada, N.; Miyahara, R.; Nedsuwan, S.; Imasanguan, W.; Kantipong, P.; Chaiyasirinroje, B.; et al. A novel Ancestral Beijing sublineage of Mycobacterium tuberculosis suggests the transition site to Modern Beijing sublineages. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, K.E.; McAdam, P.; Thai, P.V.K.; Thuong, N.T.T.; Ha, D.T.M.; Lan, N.N.; Lan, N.H.; Nhu, N.T.Q.; Hai, H.T.; Ha, V.T.N.; et al. Frequent transmission of the Mycobacterium tuberculosis Beijing lineage and positive selection for EsxW Beijing variant in Vietnam. Nat. Genet. 2018, 50, 849–856. [Google Scholar] [CrossRef]
- Phelan, J.E.; Lim, D.R.; Mitarai, S.; de Sessions, P.F.; Tujan, M.A.A.; Reyes, L.T.; Medado, I.A.P.; Palparan, A.G.; Naim, A.N.M.; Jie, S.; et al. Mycobacterium tuberculosis whole genome sequencing provides insights into the Manila strain and drug-resistance mutations in the Philippines. Sci. Rep. 2019, 9, 9305. [Google Scholar] [CrossRef] [Green Version]
- Ruesen, C.; Chaidir, L.; van Laarhoven, A.; Dian, S.; Ganiem, A.R.; Nebenzahl-Guimaraes, H.; Huynen, M.A.; Alisjahbana, B.; Dutilh, B.E.; van Crevel, R. Large-scale genomic analysis shows association between homoplastic genetic variation in Mycobacterium tuberculosis genes and meningeal or pulmonary tuberculosis. BMC Genom. 2018, 19, 122. [Google Scholar] [CrossRef] [Green Version]
- Ei, P.W.; Lee, J.S.; Aung, W.W.; Yeun, K.; Korma, W.; Nyunt, W.W.; Swe, T.L.; Htwe, M.M.; Win, S.M.; Aung, S.T.; et al. Genotypes and genetic characters of Mycobacterium tuberculosis from Myanmar using three typing methods. Infect. Genet. Evol. 2019, 75, 104005. [Google Scholar] [CrossRef] [PubMed]
- San, L.L.; Aye, K.S.; Aye, T.O.N.; Shwe, M.M.; Fukushima, Y.; Gordon, S.V.; Suzuki, Y.; Nakajima, C. Erratum to “Insight into multidrug-resistant Beijing genotype Mycobacterium tuberculosis isolates in Myanmar” [Int. J. Infect. Dis. 76 (November) (2018) 109-119]. Int. J. Infect. Dis. Off. Publ. Int. Soc. Infect. Dis. 2019, 85, 214. [Google Scholar] [CrossRef] [PubMed]
- Tun, T.; Aye, K.S.; Nyunt, W.W.; Crump, J.A.; Nakajima, C.; Suzuki, Y.; Thinn, K.K.; Cook, G.M.; Aung, H.L. Genotypic diversity of Mycobacterium tuberculosis strains in Myanmar. Infect. Dis. 2017, 49, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Aung, H.L.; Tun, T.; Moradigaravand, D.; Köser, C.U.; Nyunt, W.W.; Aung, S.T.; Lwin, T.; Thinn, K.K.; Crump, J.A.; Parkhill, J.; et al. Whole-genome sequencing of multidrug-resistant Mycobacterium tuberculosis isolates from Myanmar. J. Glob. Antimicrob. Resist. 2016, 6, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Department of Population Ministry of Immigration and Population. The 2014 Myanmar Population and Housing Census, Kayin State; Department of Population Ministry of Immigration and Population: Naypyitaw, Myanmar, 2015.
- National Tuberculosis Programme. Guidelines for the Management of Multidrug-Resistant Tuberculosis (mdr-tb) in Myanmar; National Tuberculosis Programme: Naypyitaw, Myanmar, 2017.
- Tun, T. Multidrug-resistant Mycobacterium tuberculosis Strains in Myanmar Patients. Myanmar Health Sci. Res. J. 2017, 29, 51–57. [Google Scholar]
- MO BIO’s PowerSoil DNA Isolation Kit Handbook—QIAGEN. Available online: https://www.qiagen.com/be/resources/resourcedetail?id=5c00f8e4-c9f5-4544-94fa-653a5b2a6373&lang=en (accessed on 8 September 2020).
- Phelan, J. jodyphelan/TBProfiler. 2020. Available online: https://github.com/jodyphelan/TBProfiler (accessed on 30 June 2020).
- Palittapongarnpim, P.; Ajawatanawong, P.; Viratyosin, W.; Smittipat, N.; Disratthakit, A.; Mahasirimongkol, S.; Yanai, H.; Yamada, N.; Nedsuwan, S.; Imasanguan, W.; et al. Evidence for Host-Bacterial Co-evolution via Genome Sequence Analysis of 480 Thai Mycobacterium tuberculosis Lineage 1 Isolates. Sci. Rep. 2018, 8, 11597. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, X.; Li, W.; Zhang, X.; Wang, W.; Li, C. The study on the association between Beijing genotype family and drug susceptibility phenotypes of Mycobacterium tuberculosis in Beijing. Sci. Rep. 2017, 7, 15076. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Huang, Y.; Mi, L.G.; Li, Y.X.; Liu, P.Z.; Zhang, J.; Liang, H.Y.; Li, F.; Li, H.; Zhang, S.Q.; et al. There is no correlation between sublineages and drug resistance of Mycobacterium tuberculosis Beijing/W lineage clinical isolates in Xinjiang, China. Epidemiol. Infect. 2015, 143, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Besaggio, D.; Fuselli, S.; Srikummool, M.; Kampuansai, J.; Castrì, L.; Tyler-Smith, C.; Seielstad, M.; Kangwanpong, D.; Bertorelle, G. Genetic variation in Northern Thailand Hill Tribes: Origins and relationships with social structure and linguistic differences. BMC Evol. Biol. 2007, 7, S12. [Google Scholar] [CrossRef] [Green Version]
- Listman, J.B.; Malison, R.T.; Sanichwankul, K.; Ittiwut, C.; Mutirangura, A.; Gelernter, J. Southeast Asian origins of five Hill Tribe populations and correlation of genetic to linguistic relationships inferred with genome-wide SNP data. Am. J. Phys. Anthropol. 2011, 144, 300–308. [Google Scholar] [CrossRef] [Green Version]
- Maharjan, B.; Nakajima, C.; Isoda, N.; Thapa, J.; Poudel, A.; Shah, Y.; Yamaguchi, T.; Shrestha, B.; Hoffmann, H.; Avsar, K.; et al. Genetic diversity and distribution dynamics of multidrug-resistant Mycobacterium tuberculosis isolates in Nepal. Sci. Rep. 2018, 8, 16634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aung, W.W.; Ei, P.W.; Nyunt, W.W.; Swe, T.L.; Lwin, T.; Htwe, M.M.; Kim, K.J.; Lee, J.S.; Kim, C.K.; Cho, S.N.; et al. Phenotypic and Genotypic Analysis of Anti-Tuberculosis Drug Resistance in Mycobacterium tuberculosis Isolates in Myanmar. Ann. Lab. Med. 2015, 35, 494–499. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4510501/ (accessed on 30 April 2020). [CrossRef] [PubMed] [Green Version]
- Burman, W.J.; Bliven, E.E.; Cowan, L.; Bozeman, L.; Nahid, P.; Diem, L.; Vernon, A.; Tuberculosis Trials Consortium. Relapse Associated with Active Disease Caused by Beijing Strain of Mycobacterium tuberculosis. Emerg. Infect. Dis. 2009, 15, 1061–1067. [Google Scholar] [CrossRef]
- Marais, B.; Hesseling, A.; Schaaf, H.; Gie, R.; Helden, P.; Warren, R. Mycobacterium tuberculosis Transmission Is Not Related to Household Genotype in a Setting of High Endemicity. J. Clin. Microbiol. 2009, 47, 1338–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madamarandawala, P.S.; Satyanarayana, S.; Timire, C.; Yaqoob, A.; Madegedara, D.; Magana-Arachchi, D.N. Is International Travel an Emerging Issue on Transmission of Beijing Lineage Mycobacterium tuberculosis? J. Trop. Med. 2020. [Google Scholar] [CrossRef]
- Aung, W.W.; Maung, W.; Htun, A. Study of Drug Resistant cases among new pulmonary tuberculosis patients attending a tuberculosis center, Yangon, Myanmar. Southeast. Asian J. Trop. Med. Public Health 2007, 38, 7. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L1 n (%) | L2 n (%) | L3 n (%) | L4 n (%) | Total n (%) | p Value * | |
|---|---|---|---|---|---|---|
| Total | 73 | 26 | 4 | 6 | 109 | |
| Township | 0.1 | |||||
| Hpa-An | 35 (48) | 10 (38) | 3 (75) | 5 (83) | 53 (49) | |
| Kawkareik | 24 (33) | 5 (19) | 0 (0) | 1 (17) | 30 (28) | |
| Myawaddy | 14 (19) | 11 (42) | 1 (25) | 0 (0) | 26 (24) | |
| Gender | 0.02 | |||||
| Male | 47 (64) | 23 (88) | 1 (25) | 4 (67) | 75 (69) | |
| Female | 26 (36) | 3 (12) | 3 (75) | 2 (33) | 34 (31) | |
| Age | 0.07 | |||||
| Mean (SD) | 46 (15) | 43 (13) | 58 (7) | 36 (13) | 45 (14) | |
| Age group (years) | 0.15 | |||||
| <35 | 17 (23) | 8 (31) | 0 (0) | 3 (50) | 28 (26) | |
| 35−57 | 35 (48) | 14 (54) | 1 (25) | 3 (50) | 53 (49) | |
| ≥57 | 21 (29) | 4 (15) | 3 (75) | 0 (0) | 28 (26) | |
| Ethnicity | 0.43 | |||||
| Bamar | 23 (32) | 11 (42) | 2 (50) | 2 (33) | 38 (35) | |
| Kayin | 33 (45) | 13 (50) | 1 (25) | 4 (67) | 51 (47) | |
| Others | 17 (23) | 2 (8) | 1 (25) | 0 (0) | 20 (18) |
| Major Lineage | Sublineage | n | Total (109) |
|---|---|---|---|
| L1 (EAI) | L1.1.1 | 8 | 73 |
| L1.1.1.2 | 1 | ||
| L1.1.1.5 | 2 | ||
| L1.1.1.7 | 1 | ||
| L1.1.1.8 | 3 | ||
| L1.1.2.1 | 1 | ||
| L1.1.2.2 | 10 | ||
| L1.1.3 | 3 | ||
| L1.1.3.1 | 17 | ||
| L1.2.1 | 1 | ||
| L1.2.1.1 | 2 | ||
| L1.2.1.2 | 6 | ||
| L1.2.1.3 | 1 | ||
| L1.2.2 | 17 | ||
| L2 (Beijing) | L2.2.1.1_M_Pacific | 2 | 26 |
| L2.2.1_Asia_Ancestral_2 | 1 | ||
| L2.2.1_Asia_Ancestral_3 | 1 | ||
| L2.2.1_Asia_Ancestral_4 | 3 | ||
| L2.2.1_M_Asian_African_2 | 8 | ||
| L2.2.1_M_Asian_African_3 | 1 | ||
| L2.2.1_M_Bmyc22 | 1 | ||
| L2.2.1_Modern | 8 | ||
| L2.2.2 | 1 | ||
| L3 (Delhi/CAS) | L3 | 2 | 4 |
| L3.1.2 | 1 | ||
| L3.1.2.1 | 1 | ||
| L4 (Euro-American) | L4.1.2 | 1 | 6 |
| L4.3 | 1 | ||
| L4.4 | 1 | ||
| L4.5 | 1 | ||
| L4.5.3 | 1 | ||
| L4.8 | 1 |
| Zone1 (97.458–97.646) | Zone2 (97.646–97.854) | Zone3 (97.854–98.244) | Zone4 (98.244–98.650) | Total | p Value * | |
|---|---|---|---|---|---|---|
| Total | 45 | 14 | 25 | 25 | 109 | |
| Ethnicity group | <0.001 | |||||
| Bamar | 17 (37.8) | 2 (14.3) | 3 (12.0) | 16 (64.0) | 38 (34.9) | |
| Kayin | 22 (48.9) | 4 (28.6) | 18 (72.0) | 7 (28.0) | 51 (46.8) | |
| Others | 6 (13.3) | 8 (57.1) | 4 (16.0) | 2 (8.0) | 20 (18.3) | |
| Major Linage | 0.047 | |||||
| L1 | 27 (60.0) | 14 (100) | 18 (72.0) | 14 (56.0) | 73 (67.0) | |
| L2 | 10 (22.2) | 0 (0) | 6 (24.0) | 10 (40.0) | 26 (23.9) | |
| L3 | 3 (6.7) | 0 (0) | 0 (0) | 1 (4.0) | 4 (3.7) | |
| L4 | 5 (11.1) | 0 (0) | 1 (4.0) | 0 (0) | 6 (5.5) |
| L1 n (%) | L2 n (%) | L3 n (%) | L4 n (%) | Total n (%) | p Value * | |
|---|---|---|---|---|---|---|
| Total | 73 | 26 | 4 | 6 | 109 | |
| Blood in sputum | 0.9 | |||||
| No | 55 (75) | 20 (77) | 4 (100) | 5 (83) | 84 (77) | |
| Yes | 18 (25) | 6 (23) | 0 (0) | 1 (17) | 25 (23) | |
| Fever > 2 weeks | 0.17 | |||||
| No | 34 (47) | 18 (69) | 3 (75) | 4 (67) | 59 (54) | |
| Yes | 39 (53) | 8 (31) | 1 (25) | 2 (33) | 50 (46) | |
| Unexplained weight loss | 0.35 | |||||
| No | 14 (19) | 5 (19) | 2 (50) | 2 (33) | 23 (21) | |
| Yes | 59 (81) | 21 (81) | 2 (50) | 4 (67) | 86 (79) | |
| Chest Pain | 0.35 | |||||
| No | 26 (36) | 9 (35) | 3 (75) | 1 (17) | 39 (36) | |
| Yes | 47 (64) | 17 (65) | 1 (25) | 5 (83) | 70 (64) | |
| Severity of cough | 0.05 | |||||
| Mild | 23 (32) | 8 (31) | 2 (50) | 3 (50) | 36 (33) | |
| Moderate | 41 (56) | 10 (38) | 0 (0) | 3 (50) | 54 (50) | |
| Severe | 9 (12) | 8 (31) | 2 (50) | 0 (0) | 19 (17) | |
| HIV status | 0.46 | |||||
| Positive | 3 (4) | 1 (4) | 0 (0) | 1 (17) | 5 (5) | |
| Negative | 70 (96) | 25 (96) | 4 (100) | 5 (83) | 104 (95) | |
| Type of TB Patient | 0.04 | |||||
| New | 70 (96) | 20 (77) | 4 (100) | 6 (100) | 100 (92) | |
| Retreated | 3 (4) | 6 (23) | 0 (0) | 0 (0) | 9 (8) | |
| BCG | 0.86 | |||||
| No | 23 (32) | 8 (31) | 2 (50) | 1 (17) | 34 (31) | |
| Yes | 20 (27) | 10 (38) | 1 (25) | 2 (33) | 33 (30) | |
| Unknown | 30 (41) | 8 (31) | 1 (25) | 3 (50) | 42 (39) | |
| Sputum grading | 0.01 | |||||
| Scanty | 0 (0) | 2 (8) | 0 (0) | 0 (0) | 2 (2) | |
| 1 + | 10 (14) | 9 (35) | 1 (25) | 2 (33) | 22 (20) | |
| 2 + | 16 (22) | 6 (23) | 1 (25) | 3 (50) | 26 (24) | |
| 3 + | 47 (64) | 9 (35) | 2 (50) | 1 (17) | 59 (54) | |
| Site of TB | 0.81 | |||||
| Lung | 69 (95) | 24 (92) | 4 (100) | 6 (100) | 103 (94) | |
| Lung plus other | 4 (5) | 2 (8) | 0 (0) | 0 (0) | 6 (6) | |
| L1 n (%) | L2 n (%) | L3 n (%) | L4 n (%) | Total n (%) | p Value* | |
|---|---|---|---|---|---|---|
| Total | 39 | 19 | 2 | 3 | 63 | |
| Isoniazid | 0.92 | |||||
| Sensitive | 31 (79) | 14 (74) | 2 (100) | 3 (100) | 50 (79) | |
| Resistant | 8 (21) | 5 (26) | 0 (0) | 0 (0) | 13 (21) | |
| Rifampicin | 0.24 | |||||
| Sensitive | 39 (100) | 17 (89) | 2 (100) | 3 (100) | 61 (97) | |
| Resistant | 0 (0) | 2 (11) | 0 (0) | 0 (0) | 2 (3) | |
| Pyrazinamide | 1 | |||||
| Sensitive | 38 (97) | 18 (95) | 2 (100) | 3 (100) | 61 (97) | |
| Resistant | 1 (3) | 1 (5) | 0 (0) | 0 (0) | 2 (3) | |
| Ethambutol | 0.71 | |||||
| Sensitive | 37 (95) | 17 (89) | 2 (100) | 3 (100) | 59 (94) | |
| Resistant | 2 (5) | 2 (11) | 0 (0) | 0 (0) | 4 (6) | |
| Streptomycin | 0.04 | |||||
| Sensitive | 38 (97) | 14 (74) | 2 (100) | 3 (100) | 57 (90) | |
| Resistant | 1 (3) | 5 (26) | 0 (0) | 0 (0) | 6 (10) | |
| PAS | 0.38 | |||||
| Sensitive | 39 (100) | 18 (95) | 2 (100) | 3 (100) | 62 (98) | |
| Resistant | 0 (0) | 1 (5) | 0 (0) | 0 (0) | 1 (2) | |
| Ethionamide | 0.82 | |||||
| Sensitive | 35 (90) | 16 (84) | 2 (100) | 3 (100) | 56 (89) | |
| Resistant | 4 (10) | 3 (16) | 0 (0) | 0 (0) | 7 (11) | |
| At least one TB drug-resistant | 0.42 | |||||
| Sensitive | 28 (72) | 11 (58) | 2 (100) | 3 (100) | 44 (70) | |
| Resistant | 11 (28) | 8 (42) | 0 (0) | 0 (0) | 19 (30) |
| Sub lineage | Rif | INH | PZA | ETB | STM | FQ | AM | ETO | PAS | CS |
|---|---|---|---|---|---|---|---|---|---|---|
| 2_2_1_M_ Asian_ African_2 | rpoB_p.Ser450Leu | katG_p.Ser315Thr | pncA_p.Gly97Asp | embB_p.Met306Val | rpsL_p.Lys43Arg | - | - | - | - | - |
| 2_2_2 | rpoB_p.His445Asp | katG_p.Ser315Thr | - | embB_p.Met306Ile | rpsL_p.Lys43Arg | - | - | - | - | - |
| 1_1_2_2 | - | katG_p.Ser315Thr | - | embB_p.Asp328Gly | rrs_r.514a > c | - | - | - | - | - |
| 2_2_1_Modern | - | katG_p.Ser315Thr | - | - | rrs_r.517c > t | - | - | - | - | - |
| 1_2_1 | - | katG_p.Ser315Thr | - | - | - | - | - | fabG1_c.-15C > T | - | - |
| 2_2_1_M_ Asian_ African_2 | - | fabG1_c.-15C > T | - | - | - | - | - | fabG1_c.-15C > T | - | - |
| 1_1_3_1 | - | fabG1_c.-15C > T | - | - | - | - | - | fabG1_c.-15C > T | - | - |
| 2_2_1_Modern | - | katG_p.Ser315Thr | - | - | rpsL_p.Lys43Arg | - | - | - | - | - |
| 1_1_2_2 | - | - | panD_p.Ile49Val | - | - | - | - | ethA_c.1054_1054del | - | - |
| 2_2_1_Modern | - | - | - | - | rpsL_p.Lys43Arg | - | - | ethR_p.Ala95Thr | - | - |
| 1_1_1_8 | - | katG_p.Ser315Thr | - | - | - | - | - | - | - | - |
| 1_1_3 | - | katG_p.Ser315Thr | - | - | - | - | - | - | - | - |
| 1_1_3_1 | - | katG_p.Ser315Thr | - | - | - | - | - | - | - | - |
| 1_2_1_2 | - | katG_p.Ser315Thr | - | - | - | - | - | - | - | - |
| 1_2_2 | - | - | - | - | - | - | - | ethA_c.341_341del | - | - |
| 1_1_1 | - | ahpC_c.-52C > T | - | - | - | - | - | - | - | - |
| 2_2_1_Asia_ Ancestral_3 | - | - | - | - | - | - | - | - | folC_p.Thr20Pro | - |
| 2_2_1_M_ Asian_ African_2 | - | - | - | - | - | - | - | ethA_c.24_25insC | - | - |
| 1_2_2 | - | - | - | embB_p.Phe285Leu | - | - | - | - | - | - |
| Our Study n (%) | Yangon n (%) | Thailand n (%) | Philippines n (%) | Nepal n (%) | |
|---|---|---|---|---|---|
| Lineage | 109 | 72 | 1170 | 178 | 498 |
| L1 | 73 (67%) | 9 (13%) | 480 (41%) | 143 (80%) | 32 (6%) |
| L2 | 26 (23%) | 55 (76%) | 521 (45%) | 2 (1%) | 241 (48%) |
| L3 | 4 (4%) | 4 (5%) | 11 (1%) | 0 | 153 (32%) |
| L4 | 6 (6%) | 4 (5%) | 158 (13%) | 33 (19%) | 72 (14%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maung, H.M.W.; Palittapongarnpim, P.; Aung, H.L.; Surachat, K.; Nyunt, W.W.; Chongsuvivatwong, V. Geno-Spatial Distribution of Mycobacterium Tuberculosis and Drug Resistance Profiles in Myanmar–Thai Border Area. Trop. Med. Infect. Dis. 2020, 5, 153. https://doi.org/10.3390/tropicalmed5040153
Maung HMW, Palittapongarnpim P, Aung HL, Surachat K, Nyunt WW, Chongsuvivatwong V. Geno-Spatial Distribution of Mycobacterium Tuberculosis and Drug Resistance Profiles in Myanmar–Thai Border Area. Tropical Medicine and Infectious Disease. 2020; 5(4):153. https://doi.org/10.3390/tropicalmed5040153
Chicago/Turabian StyleMaung, Htet Myat Win, Prasit Palittapongarnpim, Htin Lin Aung, Komwit Surachat, Wint Wint Nyunt, and Virasakdi Chongsuvivatwong. 2020. "Geno-Spatial Distribution of Mycobacterium Tuberculosis and Drug Resistance Profiles in Myanmar–Thai Border Area" Tropical Medicine and Infectious Disease 5, no. 4: 153. https://doi.org/10.3390/tropicalmed5040153