
Bayesian Network Analysis of Lysine Biosynthesis Pathway in Rice



Abstract
:1. Introduction
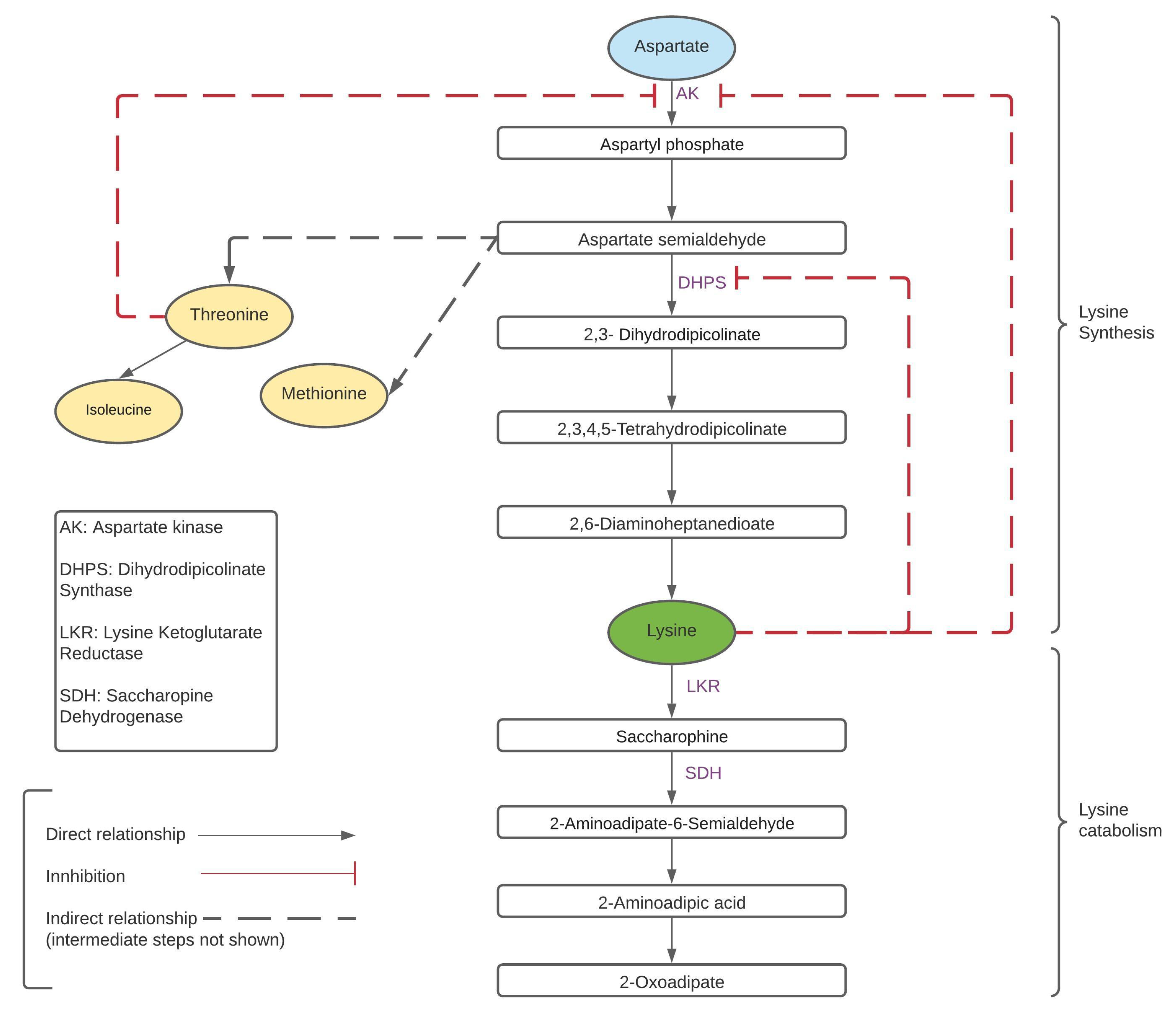
1.1. Background
1.2. Lysine Content in Rice
2. Materials and Methods
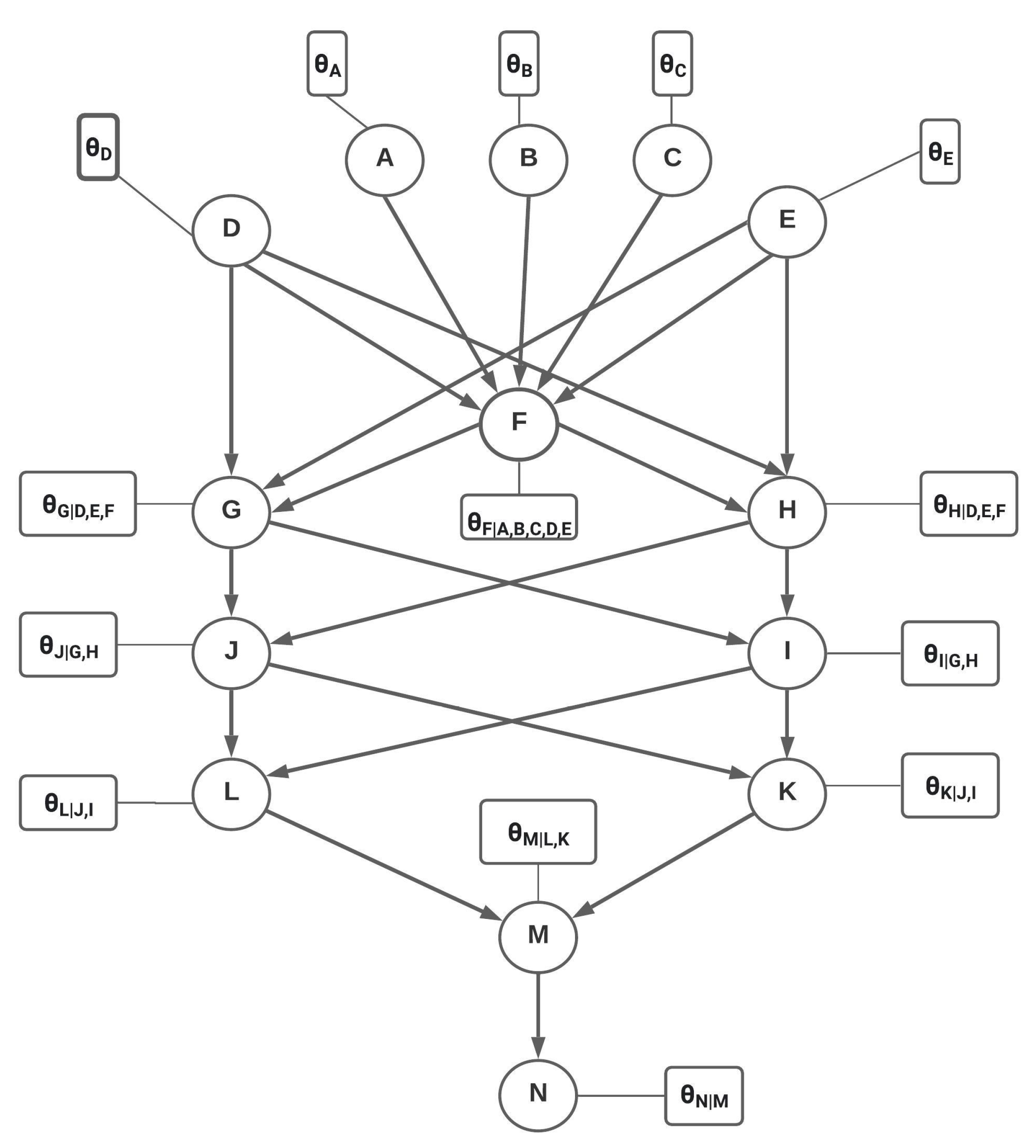
2.1. Bayesian Network Modeling
2.2. Parameter Estimation
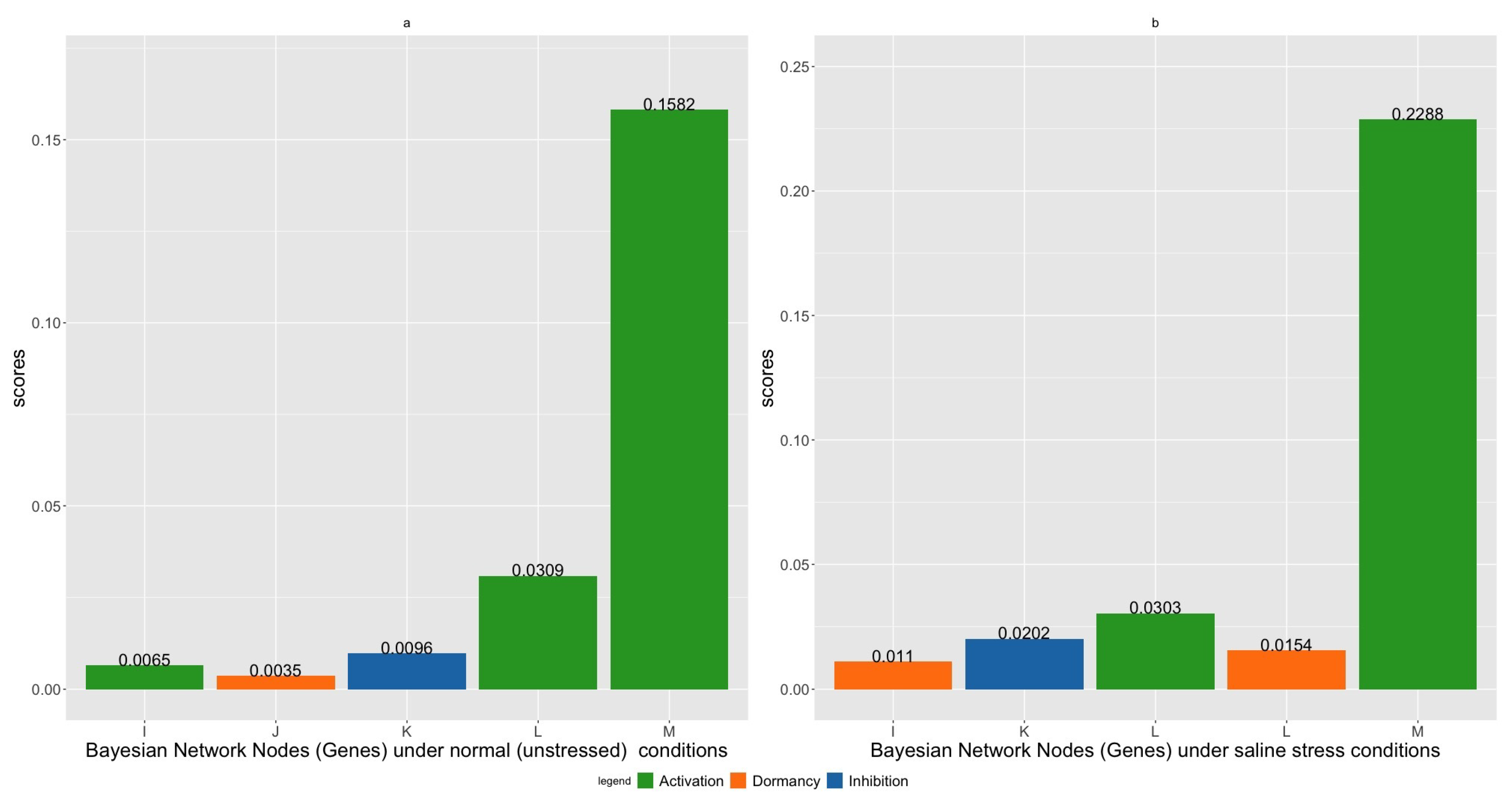
2.3. Gene Intervention Simulations
| Algorithm 1: Likelihood-Weighting Algorithm |
 |
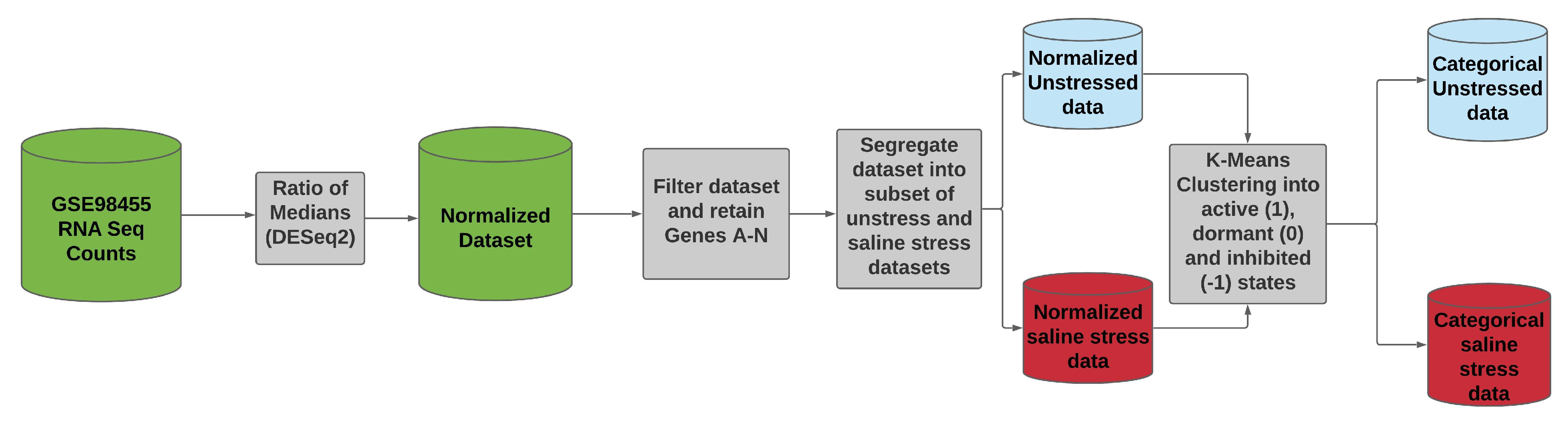
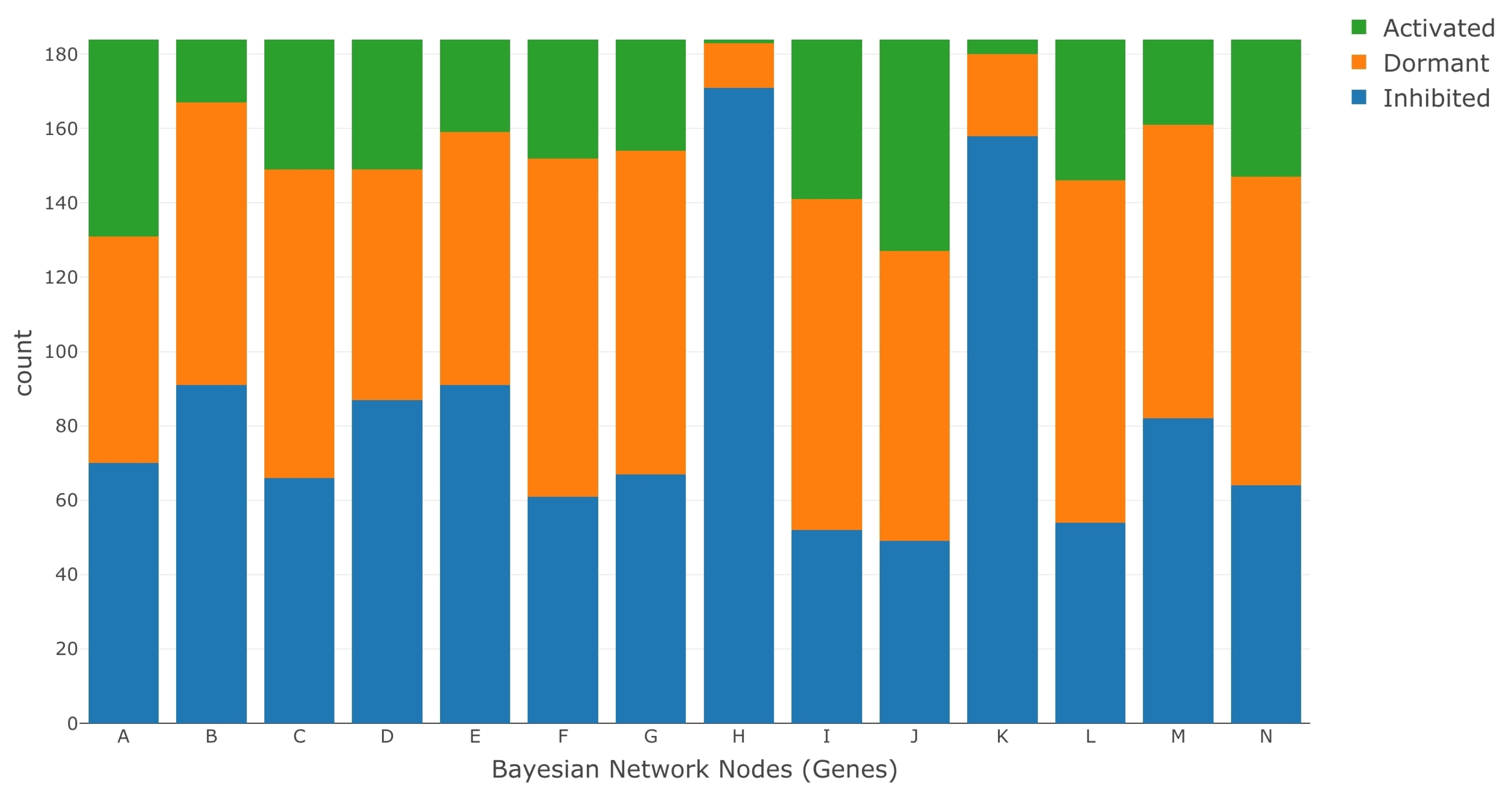
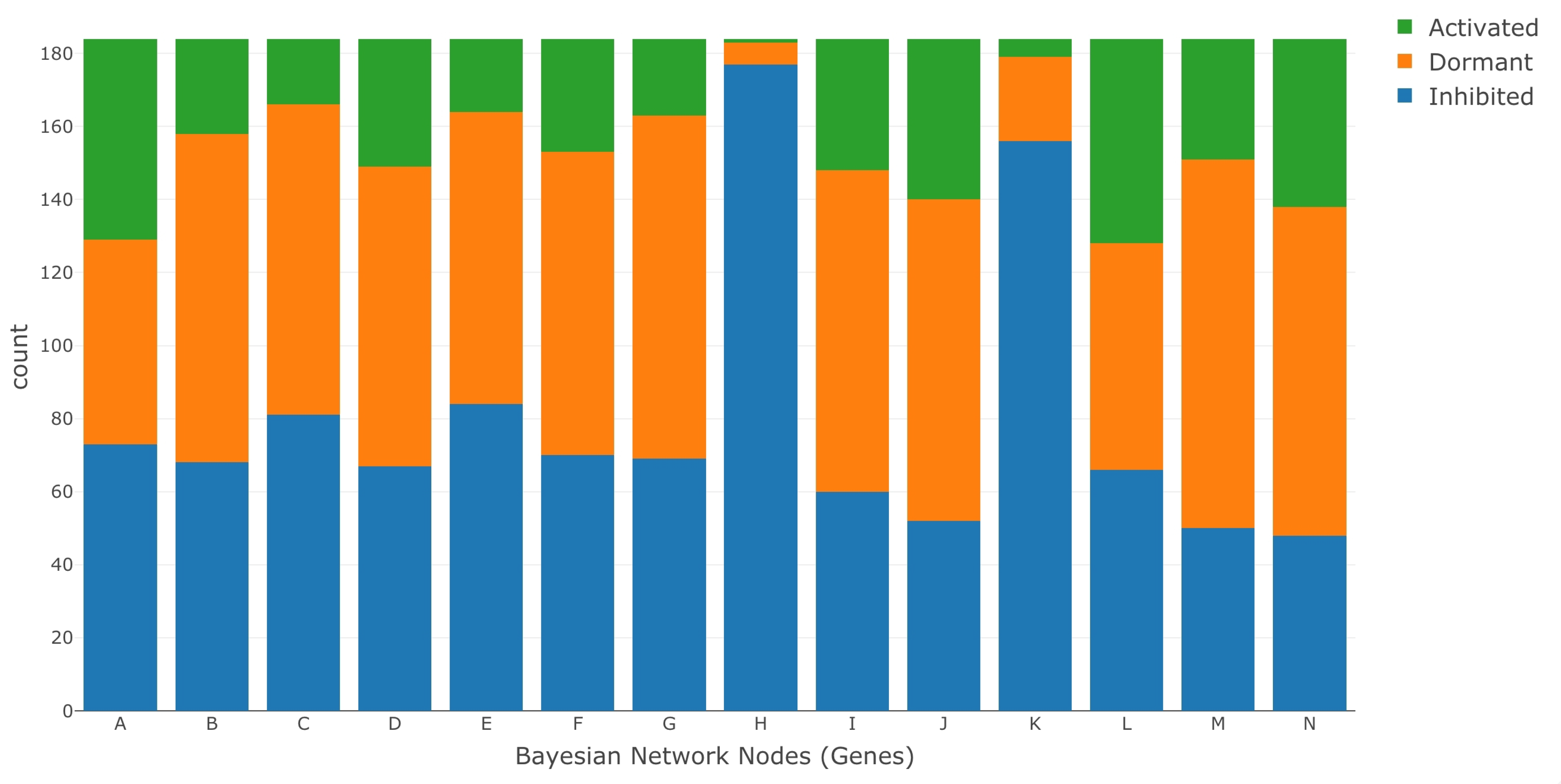
2.4. Data Set
- The entire data set was normalized using the ratio of medians methods.
- We selected the data for the genes A–N, as these were the genes in the BN model. We identified the data for each of the genes by mapping their data set IDs to their respective MSU IDs. This reduced our data set to a size of 14 rows (Gene A–N) and 368 columns.
- We further segregated the normalized data set based on saline stress and normal conditions. Since the number of columns for saline stress and normal conditions were the same, each of the resulting data set had 14 rows and 184 columns.
- We ran K-means clustering separately on both the saline stress and normal conditions data set to convert them from normalized to categorical values. The clustering process categorized the data in both the data sets into the following values 1 (active), 0 (dormant), and −1 (inhibited). The low expression values were categorized to the value of −1, the high expression values were categorized to the value of 1, and the remaining expression values in the middle were categorized to a value of 0.
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| The following abbreviations are used in this manuscript: | |
| LKR | Lysine ketoglutarate reductase |
| SDH | Saccharopine dehydrogenase |
| DHPS | Dihydrodipicolinate synthase |
| AK | Aspartate kinase |
| GRN | Gene regulatory network |
| GMO | Genetically modified organisms |
| MSU | Michigan State University |
| TF | Transcription factor |
| BN | Bayesian network |
| PGM | Probabilistic graphical model |
| LPD | Local probability distribution |
| i.i.d | Independent and identically distributed |
| LW | Likelihood weighting |
References
- Alberts, B.; Bray, D.; Hopkin, K.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Essential Cell Biology, 3rd ed.; Garland Science: New York, NY, USA, 2010; pp. 119–122. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The shape and structure of protein. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Lopez, M.; Mohiuddin, S. Biochemistry, Essential Amino Acids; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- D’Mello, J.P.F. Amino acids as multifunctional molecules. In Amino Acids in Animal Nutrition, 2nd ed.; CABI Publishing: Cambridge, MA, USA, 2003; p. 2. [Google Scholar]
- Hoffman, J.; Falvo, M. Protein-Which is Best? J. Sports Sci. Med. 2004, 3, 118. [Google Scholar]
- Tien Lea, D.; Duc Chua, H.; Quynh Lea, N. Improving Nutritional Quality of Plant Proteins Through Genetic Engineering. Curr. Genom. 2016, 17, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Stencel, C.; Dobbins, C. Report Offers New Eating and Physical Activity Targets To Reduce Chronic Disease Risk. 2002. Available online: https://www.nationalacademies.org/news/2002/09/report-offers-new-eating-and-physical-activity-targets-to-reduce-chronic-disease-risk (accessed on 10 May 2021).
- Zha, Y.; Qian, Q. Protein Nutrition and Malnutrition in CKD and ESRD. Nutrients 2017, 9, 208. [Google Scholar] [CrossRef]
- National Research Council. Recommended Dietary Allowances, 10th ed.; National Academies Press: Washington, DC, USA, 1989. [Google Scholar] [CrossRef]
- Titchenal, A.; Hara, S.; Arceo Caacbay, N.; Meinke-Lau, W.; Yang, Y.Y.; Ksinoa Fialkowski Revilla, M.; Draper, J.; Langfelder, G.; Gibby, C.; Nicole Chun, C.; et al. Human Nutrition, 2020th ed.; University of Hawaii at Manoa Food Science and Human Nutrition Program: Honolulu, HI, USA, 2020; pp. 395–402. [Google Scholar]
- Henchion, M.; Hayes, M.; Mullen, A.; Fenelon, M.; Tiwari, B. Future Protein Supply and Demand: Strategies and Factors Influencing a Sustainable Equilibrium. Foods 2017, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Vasileska, A.; Rechkoska, G. Global and Regional Food Consumption Patterns and Trends. Procedia Soc. Behav. Sci. 2012, 44, 363–369. [Google Scholar] [CrossRef] [Green Version]
- Berrazaga, I.; Micard, V.; Gueugneau, M.; Walrand, S. The Role of the Anabolic Properties of Plant- versus Animal-Based Protein Sources in Supporting Muscle Mass Maintenance: A Critical Review. Nutrients 2019, 11, 1825. [Google Scholar] [CrossRef] [Green Version]
- de Gavelle, E.; Huneau, J.F.; Bianchi, C.; Verger, E.; Mariotti, F. Protein Adequacy Is Primarily a Matter of Protein Quantity, Not Quality: Modeling an Increase in Plant:Animal Protein Ratio in French Adults. Nutrients 2017, 9, 1333. [Google Scholar] [CrossRef] [Green Version]
- Abete, I.; Romaguera, D.; Vieira, A.R.; Lopez de Munain, A.; Norat, T. Association between total, processed, red and white meat consumption and all-cause, CVD and IHD mortality: A meta-analysis of cohort studies. Br. J. Nutr. 2014, 112, 762–775. [Google Scholar] [CrossRef]
- Demeyer, D.; Mertens, B.; De Smet, S.; Ulens, M. Mechanisms Linking Colorectal Cancer to the Consumption of (Processed) Red Meat: A Review. Crit. Rev. Food Sci. Nutr. 2016, 56, 2747–2766. [Google Scholar] [CrossRef] [Green Version]
- Malik, V.S.; Li, Y.; Tobias, D.K.; Pan, A.; Hu, F.B. Dietary Protein Intake and Risk of Type 2 Diabetes in US Men and Women. Am. J. Epidemiol. 2016, 183, 715–728. [Google Scholar] [CrossRef]
- The Food and Agriculture Organization of the United Nations. Livestock Solutions for Climate Change; Technical Report; United Nations: New York, NY, USA, 2017. [Google Scholar]
- United Nations. World Population Projected to Reach 9.8 Billion in 2050, and 11.2 Billion in 2100; United Nations: New York, NY, USA, 2017. [Google Scholar]
- Day, L. Proteins from land plants–Potential resources for human nutrition and food security. Trends Food Sci. Technol. 2013, 32, 25–42. [Google Scholar] [CrossRef]
- Rosegrant, M.W.; Leach, N.; Gerpacio, R.V. Alternative futures for world cereal and meat consumption. Proc. Nutr. Soc. 1999, 58, 219–234. [Google Scholar] [CrossRef] [Green Version]
- Millward, D.J.; Jackson, A.A. Protein/energy ratios of current diets in developed and developing countries compared with a safe protein/energy ratio: Implications for recommended protein and amino acid intakes. Public Health Nutr. 2004, 7, 387–405. [Google Scholar] [CrossRef] [Green Version]
- Kusano, M.; Yang, Z.; Okazaki, Y.; Nakabayashi, R.; Fukushima, A.; Saito, K. Using Metabolomic Approaches to Explore Chemical Diversity in Rice. Mol. Plant 2015, 8, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Galili, G.; Amir, R. Fortifying plants with the essential amino acids lysine and methionine to improve nutritional quality. Plant Biotechnol. J. 2013, 11. [Google Scholar] [CrossRef]
- Wang, W.; Galili, G. Transgenic high-lysine rice–a realistic solution to malnutrition? J. Exp. Bot. 2016, 67. [Google Scholar] [CrossRef] [Green Version]
- Galili, G.; Karchi, H.; Shaul, O.; Perl, A.; Cahana, A.; Tzchori, I.B.T.; Zhu, X.Z.; Galili, S. Production of transgenic plants containing elevated levels of lysine and threonine. Biochem. Soc. Trans. 1994, 22. [Google Scholar] [CrossRef] [Green Version]
- Grigg, D. The pattern of world protein consumption. Geoforum 1995, 26. [Google Scholar] [CrossRef]
- Juliano, B.O. The Food and Agriculture Organization of the United Nations. World rice production compared to other cereals. In Rice in Human Nutrition; International Rice Research Institute of the United Nations: Rome, Italy, 1993. [Google Scholar]
- Muthayya, S.; Sugimoto, J.D.; Montgomery, S.; Maberly, G.F. An overview of global rice production, supply, trade, and consumption. Ann. N. Y. Acad. Sci. 2014, 1324, 7–14. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Takaiwa, F. Differences in Transcriptional Regulatory Mechanisms Functioning for Free Lysine Content and Seed Storage Protein Accumulation in Rice Grain. Plant Cell Physiol. 2010, 51, 1964–1974. [Google Scholar] [CrossRef] [Green Version]
- Frizzi, A.; Huang, S.; Gilbertson, L.A.; Armstrong, T.A.; Luethy, M.H.; Malvar, T.M. Modifying lysine biosynthesis and catabolism in corn with a single bifunctional expression/silencing transgene cassette. Plant Biotechnol. J. 2007. [Google Scholar] [CrossRef] [PubMed]
- Arruda, P.; Barreto, P. Lysine Catabolism Through the Saccharopine Pathway: Enzymes and Intermediates Involved in Plant Responses to Abiotic and Biotic Stress. Front. Plant Sci. 2020, 11, 587. [Google Scholar] [CrossRef]
- Long, X.; Liu, Q.; Chan, M.; Wang, Q.; Sun, S.S.M. Metabolic engineering and profiling of rice with increased lysine. Plant Biotechnol. J. 2013, 11, 490–501. [Google Scholar] [CrossRef]
- Arruda, P.; Kemper, E.L.; Papes, F.; Leite, A. Regulation of lysine catabolism in higher plants. Trends Plant Sci. 2000, 5, 324–330. [Google Scholar] [CrossRef]
- Yang, Q.Q.; Zhang, C.Q.; Chan, M.L.; Zhao, D.S.; Chen, J.Z.; Wang, Q.; Li, Q.F.; Yu, H.X.; Gu, M.H.; Sun, S.S.M.; et al. Biofortification of rice with the essential amino acid lysine: Molecular characterization, nutritional evaluation, and field performance. J. Exp. Bot. 2016, 67, 4285–4296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Galili, G. Increased Lysine Synthesis Coupled with a Knockout of Its Catabolism Synergistically Boosts Lysine Content and Also Transregulates the Metabolism of Other Amino Acids in Arabidopsis Seeds. Plant Cell 2003, 15, 845–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelovici, R.; Fait, A.; Fernie, A.R.; Galili, G. A seed high-lysine trait is negatively associated with the TCA cycle and slows down Arabidopsis seed germination. New Phytol. 2011, 189, 148–159. [Google Scholar] [CrossRef]
- Tzchori, I.B.T.; Perl, A.; Galili, G. Lysine and threonine metabolism are subject to complex patterns of regulation in Arabidopsis. Plant Mol. Biol. 1996, 32, 727–734. [Google Scholar] [CrossRef]
- Rappe, M. CRISPR Plants: New Non-GMO Method to Edit Plants; North Carolina State University: Raleigh, NC, USA, 2020. [Google Scholar]
- Shew, A.M.; Nalley, L.L.; Snell, H.A.; Nayga, R.M.; Dixon, B.L. CRISPR versus GMOs: Public acceptance and valuation. Glob. Food Secur. 2018, 19, 71–80. [Google Scholar] [CrossRef]
- Rastogi, K.; Ibarra, O.; Molina, M.; Faion-Molina, M.; Thomson, M.; Septiningsih, E.M. Using CRISPR/Cas9 Genome Editing to Increase Lysine Levels in Rice. In Proceedings of the ASA-CSSA-SSSA International Annual Meeting, San Antonio, TX, USA, 14 November 2019. [Google Scholar]
- Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, A.; Venkatasubramani, P.S.; Datta, A. Bayesian modeling of plant drought resistance pathway. BMC Plant Biol. 2019, 19. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, A.; Zhou, L.; He, P.; Datta, A. Detecting Drought Regulators using Stochastic Inference in Bayesian Networks. Manuscript submitted for publication. [CrossRef]
- Sheng, M.; Tang, M.; Chen, H.; Yang, B.; Zhang, F.; Huang, Y. Influence of arbuscular mycorrhizae on photosynthesis and water status of maize plants under salt stress. Mycorrhiza 2008, 18, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Tisarum, R.; Theerawitaya, C.; Samphumphuang, T.; Polispitak, K.; Thongpoem, P.; Singh, H.P.; Cha-um, S. Alleviation of Salt Stress in Upland Rice (Oryza sativa L. ssp. indica cv. Leum Pua) Using Arbuscular Mycorrhizal Fungi Inoculation. Front. Plant Sci. 2020, 11, 348. [Google Scholar] [CrossRef] [Green Version]
- Reddy, I.N.B.L.; Kim, B.K.; Yoon, I.S.; Kim, K.H.; Kwon, T.R. Salt Tolerance in Rice: Focus on Mechanisms and Approaches. Rice Sci. 2017, 24, 123–144. [Google Scholar] [CrossRef]
- Kakar, N.; Jumaa, S.H.; Redoña, E.D.; Warburton, M.L.; Reddy, K.R. Evaluating rice for salinity using pot-culture provides a systematic tolerance assessment at the seedling stage. Rice 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, V.; Mankar, S.P.; Muthukumar, C.; Divahar, P.; Bharathi, A.; Thomas, H.B.; Rajurkar, A.; Sellamuthu, R.; Poornima, R.; Senthivel, S.; et al. Genome-Wide Consistent Molecular Markers Associated with Phenology, Plant Production and Root Traits in Diverse Rice (Oryza sativa L.) Accessions under Drought in Rainfed Target Populations of the Environment. Curr. Sci. 2018, 114, 329–340. [Google Scholar] [CrossRef]
- Razzaque, S.; Elias, S.M.; Haque, T.; Biswas, S.; Jewel, G.M.N.A.; Rahman, S.; Weng, X.; Ismail, A.M.; Walia, H.; Juenger, T.E.; et al. Gene Expression analysis associated with salt stress in a reciprocally crossed rice population. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Stewart, G.; Larher, F. Accumulation of Amino Acids and Related Compounds in Relation to Environmental Stress. Amino Acids Deriv. 1980, 609–635. [Google Scholar] [CrossRef]
- Ali, Q.; Athar, H.U.R.; Haider, M.Z.; Shahid, S.; Aslam, N.; Shehzad, F.; Naseem, J.; Ashraf, R.; Ali, A.; Hussain, S.M.; et al. Role of Amino Acids in Improving Abiotic Stress Tolerance to Plants. Plant Toler. Environ. Stress 2019, 175–204. [Google Scholar] [CrossRef]
- Wang, M.; Liu, C.; Li, S.; Zhu, D.; Zhao, Q.; Yu, J. Improved Nutritive Quality and Salt Resistance in Transgenic Maize by Simultaneously Overexpression of a Natural Lysine-Rich Protein Gene, SBgLR, and an ERF Transcription Factor Gene, TSRF1. Int. J. Mol. Sci. 2013, 14, 9459–9474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeedipour, S. Stress-induced changes in the free amino acid composition of two wheat cultivars with difference in drought resistance. Afr. J. Biotechnol. 2012, 11, 9559–9565. [Google Scholar] [CrossRef]
- Jackson, C.A.; Castro, D.M.; Saldi, G.A.; Bonneau, R.; Gresham, D. Gene regulatory network reconstruction using single-cell RNA sequencing of barcoded genotypes in diverse environments. eLife 2020, 9. [Google Scholar] [CrossRef]
- Davidson, E.H.; Erwin, D.H. Gene Regulatory Networks and the Evolution of Animal Body Plans. Science 2006, 311, 796–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kærn, M.; Blake, W.J.; Collins, J. The Engineering of Gene Regulatory Networks. Annu. Rev. Biomed. Eng. 2003, 5, 179–206. [Google Scholar] [CrossRef] [Green Version]
- Bonnaffoux, A.; Herbach, U.; Richard, A.; Guillemin, A.; Gonin-Giraud, S.; Gros, P.A.; Gandrillon, O. WASABI: A dynamic iterative framework for gene regulatory network inference. BMC Bioinform. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Dinneny, J.R. Q&A: How do gene regulatory networks control environmental responses in plants? BMC Biol. 2018, 16. [Google Scholar] [CrossRef]
- Emmert-Streib, F.; Dehmer, M.; Haibe-Kains, B. Gene regulatory networks and their applications: Understanding biological and medical problems in terms of networks. Front. Cell Dev. Biol. 2014, 2, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arshad, O.A.; Datta, A. Towards targeted combinatorial therapy design for the treatment of castration-resistant prostate cancer. BMC Bioinform. 2017, 18, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Vundavilli, H.; Datta, A.; Sima, C.; Hua, J.; Lopes, R.; Bittner, M. Targeting oncogenic mutations in colorectal cancer using cryptotanshinone. PLoS ONE 2021, 16, e0247190. [Google Scholar] [CrossRef]
- Timmermann, T.; González, B.; Ruz, G.A. Reconstruction of a gene regulatory network of the induced systemic resistance defense response in Arabidopsis using boolean networks. BMC Bioinform. 2020, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkat, P.S.; Narayanan, K.R.; Datta, A. A Bayesian Network-Based Approach to Selection of Intervention Points in the Mitogen-Activated Protein Kinase Plant Defense Response Pathway. J. Comput. Biol. 2017, 24. [Google Scholar] [CrossRef] [PubMed]
- Vijesh, N.; Chakrabarti, S.K.; Sreekumar, J. Modeling of gene regulatory networks: A review. J. Biomed. Sci. Eng. 2013, 6, 223. [Google Scholar] [CrossRef] [Green Version]
- Vundavilli, H.; Datta, A.; Sima, C.; Hua, J.; Lopes, R.; Bittner, M. Bayesian Inference Identifies Combination Therapeutic Targets in Breast Cancer. IEEE Trans. Biomed. Eng. 2019, 66. [Google Scholar] [CrossRef] [PubMed]
- Karlebach, G.; Shamir, R. Modelling and analysis of gene regulatory networks. Nat. Rev. Mol. Cell Biol. 2008, 9, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Vundavilli, H.; Datta, A.; Sima, C.; Hua, J.; Lopes, R.; Bittner, M. Cryptotanshinone Induces Cell Death in Lung Cancer by Targeting Aberrant Feedback Loops. IEEE J. Biomed. Health Inform. 2020, 24. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Datta, A.; Sima, C.; Hua, J.; Lopes, R.; Bittner, M.L. A Gaussian Mixture-Model Exploiting Pathway Knowledge for Dissecting Cancer Heterogeneity. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 17, 459–468. [Google Scholar] [CrossRef]
- Sinoquet, C.; Mourad, R. Probabilistic Graphical Models for Next-generation Genomics and Genetics. In Probabilistic Graphical Models for Genetics, Genomics, and Postgenomics; Oxford University Press: Oxford, UK, 2014; pp. 1–16. [Google Scholar] [CrossRef]
- Heckerman, D.; Breese, J. Causal independence for probability assessment and inference using Bayesian networks. IEEE Trans. Syst. Man Cybern. Part A Syst. Hum. 1996, 26. [Google Scholar] [CrossRef] [Green Version]
- Borsuk, M.E.; Stow, C.A.; Reckhow, K.H. A Bayesian network of eutrophication models for synthesis, prediction, and uncertainty analysis. Ecol. Model. 2004, 173. [Google Scholar] [CrossRef]
- Sevinc, V.; Kucuk, O.; Goltas, M. A Bayesian network model for prediction and analysis of possible forest fire causes. For. Ecol. Manag. 2020, 457, 117723. [Google Scholar] [CrossRef]
- Neapolitan, R.E. Learning Bayesian Networks; Prentice Hall: Hoboken, NJ, USA, 2004; p. 433. [Google Scholar]
- Kabli, R.; Herrmann, F.; McCall, J. A chain-model genetic algorithm for Bayesian network structure learning. In Proceedings of the 9th Annual Conference on Genetic and Evolutionary Computation-GECCO ’07, London, UK, July 2007; ACM Press: New York, NY, USA, 2007. [Google Scholar] [CrossRef]
- Scanagatta, M.; Salmerón, A.; Stella, F. A survey on Bayesian network structure learning from data. Prog. Artif. Intell. 2019, 8, 425–439. [Google Scholar] [CrossRef]
- Zhang, N.L. COMP538: Introduction to Bayesian Networks Lecture 6: Parameter Learning in Bayesian Networks. 2008. Available online: https://www.cse.ust.hk/bnbook/pdf/l06.h.pdf (accessed on 4 April 2021).
- Spiegelhalter, D. Lecture 6: Bayesian Estimation. 2016. Available online: http://www.statslab.cam.ac.uk/Dept/People/djsteaching/S1B-17-06-bayesian.pdf (accessed on 4 April 2021).
- Fan, Z.; Chin, A. Lecture 20—Bayesian Analysis. 2016. Available online: http://web.stanford.edu/class/stats200/Lecture20.pdf (accessed on 4 April 2021).
- Orlof, J.; Bloom, J. Comparison of Frequentist and Bayesian Inference. 2014. Available online: https://ocw.mit.edu/courses/mathematics/18-05-introduction-to-probability-and-statistics-spring-2014/readings/MIT18_05S14_Reading20.pdf (accessed on 4 April 2021).
- Storkey, A.J. Machine Learning and Pattern Recognition: Note on Dirichlet Multinomial. 2020. Available online: http://www.inf.ed.ac.uk/teaching/courses/mlpr/assignments/multinomial.pdf (accessed on 4 April 2021).
- Liu, H.; Wasserman, L. Bayesian Inference. In Statistical Machine Learning; Carnegie Mellon University: Pittsburgh, PA, USA, 2014; pp. 299–305. [Google Scholar]
- Alvares, D.; Armero, C.; Forte, A. What Does Objective Mean in a Dirichlet-multinomial Process? Int. Stat. Rev. 2018, 86. [Google Scholar] [CrossRef]
- Kelly, D.; Atwood, C. Finding a minimally informative Dirichlet prior distribution using least squares. Reliab. Eng. Syst. Saf. 2011, 96. [Google Scholar] [CrossRef]
- Robert, C.P. Bayesian computational tools. Annu. Rev. Stat. Its Appl. 2014, 1, 153–177. [Google Scholar] [CrossRef] [Green Version]
- Koller, D.; Friedman, F. Bayesian Parameter Estimation. In Probabilistic Graphical Models; MIT Press: Cambridge, MA, USA, 2009; pp. 738–739. [Google Scholar]
- Bielza, C.; Larrañaga, P. Bayesian networks in neuroscience: A survey. Front. Comput. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Shimony, S.E. Finding MAPs for belief networks is NP-hard. Artif. Intell. 1994, 68, 399–410. [Google Scholar] [CrossRef]
- Pearl, J. Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference, 1st ed.; Morgan Kaufmann Publishers, INC: San Francisco, CA, USA, 1988. [Google Scholar]
- Lozano-Pérez, T.; Kaelbling, K. 6.825 Techniques in Artificial Intelligence (SMA 5504); MIT OpenCourseWare: Cambridge, MA, USA, 2002. [Google Scholar]
- Guo, H.; Hsu, W. A Survey of Algorithms for Real-Time Bayesian Network Inference; Technical Report; Association for the Advancement of Artificial Intelligence: Menlo Park, CA, USA, 2002. [Google Scholar]
- Shwe, M.; Cooper, G. An empirical analysis of likelihood-weighting simulation on a large, multiply connected medical belief network. Comput. Biomed. Res. 1991, 24, 453–475. [Google Scholar] [CrossRef]
- Russell, S.; Norvig, P. Artificial Intelligence: A Modern Approach, 3rd ed.; Prentice Hall: Hoboken, NJ, USA, 2010; pp. 533–535. [Google Scholar]
- National Library of Medicine. National Center for Biotechnology Information; National Library of Medicine: Bethesda, Maryland, 1988.
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Res. 2012, 41. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15. [Google Scholar] [CrossRef] [Green Version]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17. [Google Scholar] [CrossRef] [Green Version]
- Wen, G. A Simple Process of RNA-Sequence Analyses by Hisat2, Htseq and DESeq2. In Proceedings of the 2017 International Conference on Biomedical Engineering and Bioinformatics-ICBEB, Bangkok, Thailand, September 2017; ACM Press: New York, NY, USA, 2017. [Google Scholar] [CrossRef]
- Jeong, H.H.; Liu, Z. Are HHV-6A and HHV-7 Really More Abundant in Alzheimer’s Disease? Neuron 2019, 104. [Google Scholar] [CrossRef]
- Nagarajan, R.; Scutari, M.; Lèbre, S. Bayesian Networks in R; Springer: New York, NY, USA, 2013. [Google Scholar] [CrossRef]
- Scutari, M. Learning Bayesian Networks with the bnlearn R Package. J. Stat. Softw. 2010, 35. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index | Gene Name/Alias | Intervention | Gene Name/Alias | Intervention | Score |
|---|---|---|---|---|---|
| 1 | ALD1 (Gene K) | Active | DAPF (Gene M) | Active | 0.1657 |
| 2 | ALD1 (Gene K) | Dormant | DAPF(Gene M) | Active | 0.1653 |
| 3 | AGD2 (Gene L) | Inhibited | DAPF (Gene M) | Active | 0.1639 |
| 4 | Gene A | Active | DAPF (Gene M) | Active | 0.1637 |
| 5 | Gene C | Active | DAPF (Gene M) | Active | 0.1634 |
| Index | Gene Name/Alias | Intervention | Gene Name/Alias | Intervention | Score |
|---|---|---|---|---|---|
| 1 | Gene F | Dormant | DAPF (Gene M) | Active | 0.2322 |
| 2 | ALD1 (Gene K) | Dormant | DAPF(Gene M) | Active | 0.2321 |
| 3 | Gene B | Inhibited | DAPF (Gene M) | Active | 0.2312 |
| 4 | Gene E | Inhibited | DAPF (Gene M) | Active | 0.2306 |
| 5 | Gene A | Dormant | DAPF (Gene M) | Active | 0.2305 |
| Gene Alias/Name | MSU IDs | Protein |
|---|---|---|
| Gene A | LOC_Os01g70300 | Aspartokinase 3, chloroplast precursor, putative, expressed |
| Gene B | LOC_Os03g63330 | Aspartokinase, chloroplast precursor, putative, expressed |
| Gene C | LOC_Os07g20544 | Aspartokinase, chloroplast precursor, putative, expressed |
| Gene E | LOC_Os09g12290 | Bifunctional aspartokinase/homoserine dehydrogenase, chloroplast precursor, putative, expressed |
| Gene F | LOC_Os03g55280 | Semialdehyde dehydrogenase, NAD binding domain containing protein, putative, expressed |
| Gene I/DAPB1 | LOC_Os02g24020 | Dihydrodipicolinate reductase, putative, expressed |
| Gene J/ DAPB2 | LOC_Os03g14120 | Dihydrodipicolinate reductase, putative, expressed |
| Gene K/ALD1 | LOC_Os03g09910 | Aminotransferase, classes I and II, domain containing protein, expressed |
| Gene L/AGD2 | LOC_Os03g18810 | Aminotransferase, classes I and II, domain containing protein, expressed |
| Gene M/DAPF | LOC_Os12g37960 | Diaminopimelate epimerase, chloroplast precursor, putative, expressed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lahiri, A.; Rastogi, K.; Datta, A.; Septiningsih, E.M. Bayesian Network Analysis of Lysine Biosynthesis Pathway in Rice. Inventions 2021, 6, 37. https://doi.org/10.3390/inventions6020037
Lahiri A, Rastogi K, Datta A, Septiningsih EM. Bayesian Network Analysis of Lysine Biosynthesis Pathway in Rice. Inventions. 2021; 6(2):37. https://doi.org/10.3390/inventions6020037
Chicago/Turabian StyleLahiri, Aditya, Khushboo Rastogi, Aniruddha Datta, and Endang M. Septiningsih. 2021. "Bayesian Network Analysis of Lysine Biosynthesis Pathway in Rice" Inventions 6, no. 2: 37. https://doi.org/10.3390/inventions6020037