
Improvement and Validation of a Genomic DNA Extraction Method for Human Breastmilk

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Milk Sampling and Sample Processing
2.2. Bacteria and Yeast Growth Conditions
2.3. Cell Counting and Preparation of the Mock Community
2.4. DNA Extraction Methods
2.4.1. Zymo Method (ZYMO)
2.4.2. Guanidinium Thiocyanate Method (GTC)
2.4.3. Cetyltrimethylammonium Bromide (CTAB)-Based Method with Doubled Phenol Extraction (CTAB-2PH)
2.4.4. CTAB Standardized for Human Milk Samples (CTAB-STD)
2.5. Spectrophotometric Analyses of DNA
2.6. PCR Amplification
2.7. Agarose Gel Electrophoresis
2.8. Statistical Analysis
3. Results
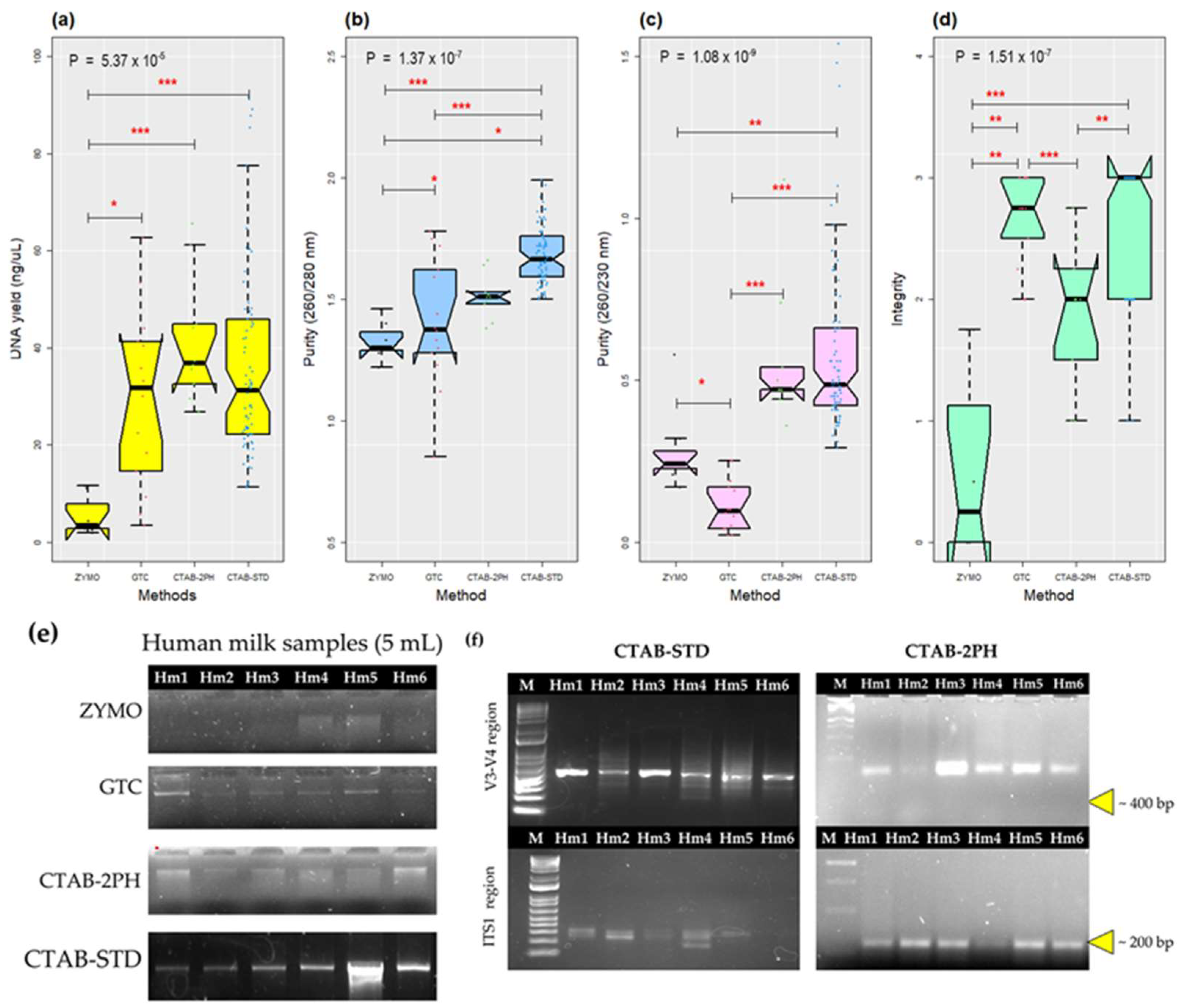
3.1. The Improvement of the CTAB-STD Method Increased the Quantity and Quality of the gDNA
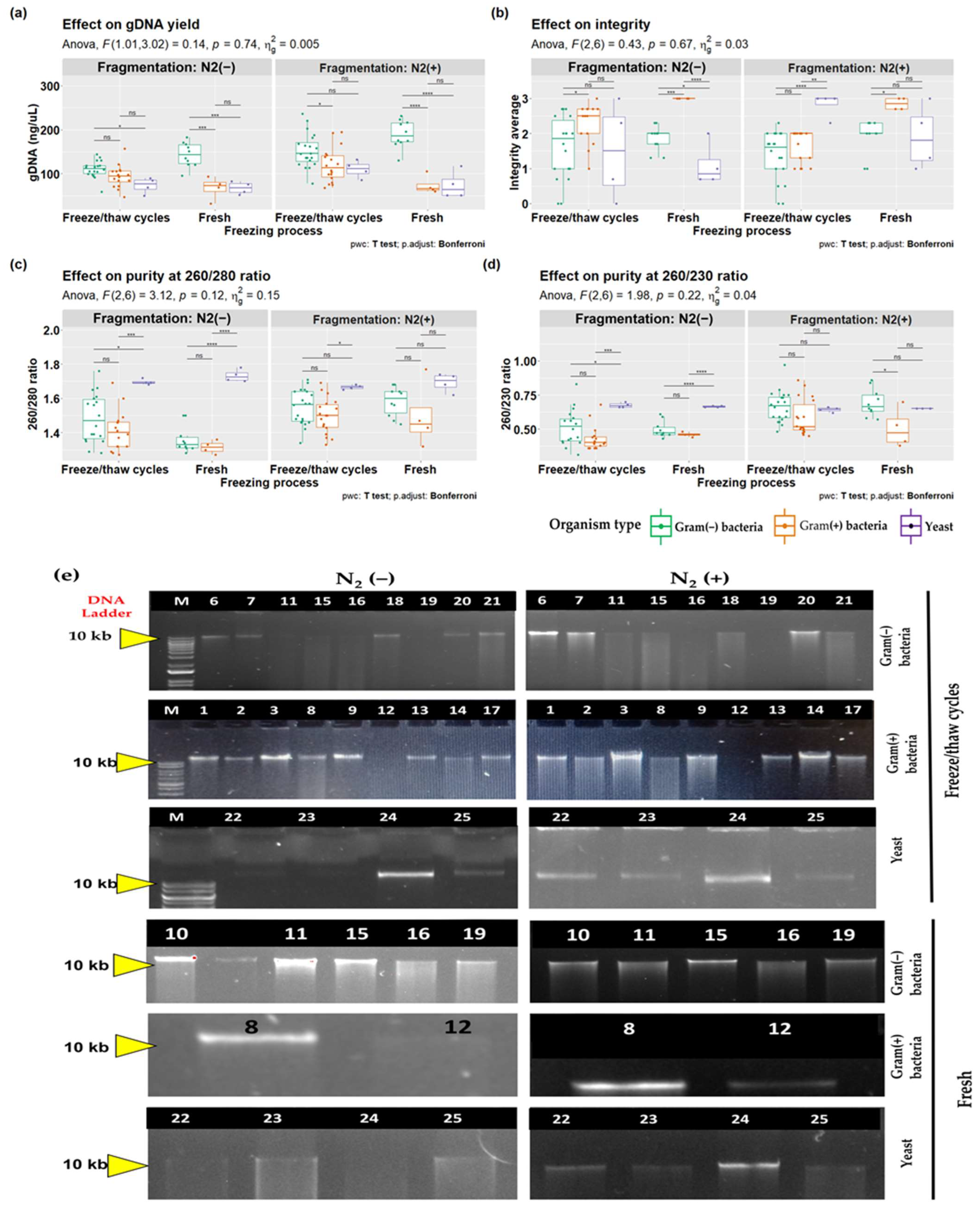
3.2. CTAB-STD Method Can Extract a High Quantity and Quality of gDNA from Fungi, Gram-Positive, and Gram-Negative Bacteria
3.3. Effects of Mechanical Fragmentation on DNA Extraction from Different Cell Types
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vieites, J.M.; Guazzaroni, M.E.; Beloqui, A.; Golyshin, P.N.; Ferrer, M. Molecular methods to study complex microbial communities. In Metagenomics Methods and Protocols; Wolfgang, R.S., Rolf, D., Eds.; Springer Protocols: London, UK, 2010; Volume 668, pp. 1–37. ISBN 978-1-60761-822-5. [Google Scholar]
- Donovan, S.M. Human Milk Proteins: Composition and Physiological Significance. Nestle Nutr. Inst. Workshop Ser. 2019, 90, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, L.; Langa, S.; Martín, V.; Maldonado, A.; Jiménez, E.; Martín, R.; Rodríguez, J.M. The human milk microbiota: Origin and potential roles in health and disease. Pharmacol. Res. 2013, 69, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Boix-Amorós, A.; Collado, M.C.; Mira, A. Relationship between milk microbiota, bacterial load, macronutrients, and human cells during lactation. Front. Microbiol. 2016, 7, 492. [Google Scholar] [CrossRef] [Green Version]
- Fitzstevens, J.L.; Smith, K.C.; Hagadorn, J.I.; Caimano, M.J.; Matson, A.P.; Brownell, E.A. Systematic review of the human milk microbiota. Nutr. Clin. Pract. 2017, 32, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, E.; De Andrés, J.; Manrique, M.; Pareja-Tobes, P.; Tobes, R.; Martínez-Blanch, J.F.; Codoñer, F.M.; Ramón, D.; Fernández, L.; Rodríguez, J.M. Metagenomic analysis of milk of healthy and mastitis-suffering women. J. Hum. Lact. 2015, 31, 406–415. [Google Scholar] [CrossRef]
- Li, S.-W.; Watanabe, K.; Hsu, C.-C.; Chao, S.-H.; Yang, Z.-H.; Lin, Y.-J.; Chen, C.-C.; Cao, Y.-M.; Huang, H.-C.; Chang, C.-H.; et al. Bacterial Composition and Diversity in Breast Milk Samples from Mothers Living in Taiwan and Mainland China. Front. Microbiol. 2017, 8, 965. [Google Scholar] [CrossRef]
- Boix-Amorós, A.; Martinez-Costa, C.; Querol, A.; Collado, M.C.; Mira, A. Multiple Approaches Detect the Presence of Fungi in Human Breastmilk Samples from Healthy Mothers. Sci. Rep. 2017, 7, 13016. [Google Scholar] [CrossRef] [Green Version]
- Boix-Amorós, A.; Puente-Sánchez, F.; du Toit, E.; Linderborg, K.M.; Zhang, Y.; Yang, B.; Salminen, S.; Isolauri, E.; Tamames, J.; Mira, A.; et al. Mycobiome profiles in breast milk from healthy women depend on mode of delivery, geographic location, and interaction with bacteria. Appl. Environ. Microbiol. 2019, 85, e02994-18. [Google Scholar] [CrossRef] [Green Version]
- Moossavi, S.; Fehr, K.; Derakhshani, H.; Sbihi, H.; Robertson, B.; Bode, L.; Brook, J.; Turvey, S.E.; Moraes, T.J.; Becker, A.B.; et al. Human milk fungi: Environmental determinants and inter-kingdom associations with milk bacteria in the CHILD Cohort Study. BMC Microbiol. 2020, 20, 146. [Google Scholar] [CrossRef]
- Dinleyici, M.; Pérez-Brocal, V.; Arslanoglu, S.; Aydemir, O.; Ozumut, S.S.; Tekin, N.; Vandenplas, Y.; Moya, A.; Dinleyici, E.C. Human milk mycobiota composition: Relationship with gestational age, delivery mode, and birth weight. Benef. Microbes 2020, 11, 151–162. [Google Scholar] [CrossRef]
- Togo, A.H.; Grine, G.; Khelaifia, S.; des Robert, C.; Brevaut, V.; Caputo, A.; Baptiste, E.; Bonnet, M.; Levasseur, A.; Drancourt, M.; et al. Culture of Methanogenic Archaea from Human Colostrum and Milk. Sci. Rep. 2019, 9, 18653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Guglielmo, M.D.; Franke, K.; Cox, C.; Crowgey, E.L. Whole genome metagenomic analysis of the gut microbiome of differently fed infants identifies differences in microbial composition and functional genes, including an absent CRISPR/Cas9 gene in the formula-fed cohort. Hum. Microbiome J. 2019, 12, 100057. [Google Scholar] [CrossRef]
- Drago, L.; Toscano, M.; De Grandi, R.; Grossi, E.; Padovani, E.M.; Peroni, D.G. Microbiota network and mathematic microbe mutualism in colostrum and mature milk collected in two different geographic areas: Italy versus Burundi. ISME J. 2017, 11, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Parigi, S.M.; Eldh, M.; Larssen, P.; Gabrielsson, S.; Villablanca, E.J. Breast milk and solid food shaping intestinal immunity. Front. Immunol. 2015, 6, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Gallego, C.; Garcia-Mantrana, I.; Salminen, S.; Collado, M.C. The human milk microbiome and factors influencing its composition and activity. Semin. Fetal Neonatal Med. 2016, 21, 400–405. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Guan, Q.; Ye, C.; Zhang, C.; Foster, J.A.; Forney, L.J. Network analysis suggests a potentially “evil” alliance of opportunistic pathogens inhibited by a cooperative network in human milk bacterial communities. Sci. Rep. 2015, 5, 8275. [Google Scholar] [CrossRef] [Green Version]
- Sakwinska, O.; Moine, D.; Delley, M.; Combremont, S.; Rezzonico, E.; Descombes, P.; Vinyes-Pares, G.; Zhang, Y.; Wang, P.; Thakkar, S.K. Microbiota in breast milk of Chinese lactating mothers. PLoS ONE 2016, 11, e0160856. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.C.; Isolauri, E.; Laitinen, K.; Salminen, S. Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am. J. Clin. Nutr. 2008, 88, 894–899. [Google Scholar] [CrossRef] [Green Version]
- Moossavi, S.; Sepehri, S.; Robertson, B.; Bode, L.; Goruk, S.; Field, C.J.; Lix, L.M.; de Souza, R.J.; Becker, A.B.; Mandhane, P.J.; et al. Composition and Variation of the Human Milk Microbiota Are Influenced by Maternal and Early-Life Factors. Cell Host Microbe 2019, 25, 324–335.e4. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Jiang, J.; Lu, M.; Tong, W.; Zhou, R.; Li, J.; Yuan, J.; Wang, F.; Li, D. Human milk microbiota development during lactation and its relation to maternal geographic location and gestational hypertensive status. Gut Microbes 2020, 11, 1438–1449. [Google Scholar] [CrossRef]
- Burgess, A.; Shah, K.; Hough, O.; Hynynen, K. Bacterial microbiome of breast milk and child saliva from low- income Mexican-American women and children. Pediatr. Res. 2016, 15, 477–491. [Google Scholar] [CrossRef]
- Ojo-Okunola, A.; Nicol, M.; du Toit, E. Human breast milk bacteriome in health and disease. Nutrients 2018, 10, 1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodayar-Pardo, P.; Mira-Pascual, L.; Collado, M.C.; Martínez-Costa, C. Impact of lactation stage, gestational age and mode of delivery on breast milk microbiota. J. Perinatol. 2014, 34, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.; Matsumoto, A.; Rutherford, C.; Lima, H.K. Comparison of the Effectiveness of Four Commercial DNA Extraction Kits on Fresh and Frozen Human Milk Samples. Methods Protoc. 2022, 5, 63. [Google Scholar] [CrossRef]
- Lyons, K.E.; Fouhy, F.; O’ Shea, C.A.; Ryan, C.A.; Dempsey, E.M.; Ross, R.P.; Stanton, C. Effect of storage, temperature, and extraction kit on the phylogenetic composition detected in the human milk microbiota. Microbiologyopen 2021, 10, e1127. [Google Scholar] [CrossRef]
- Lackey, K.A.; Williams, J.E.; Price, W.J.; Carrothers, J.M.; Brooker, S.L.; Shafii, B.; McGuire, M.A.; McGuire, M.K. Comparison of commercially-available preservatives for maintaining the integrity of bacterial DNA in human milk. J. Microbiol. Methods 2017, 141, 73–81. [Google Scholar] [CrossRef]
- Ojo-Okunola, A.; Claassen-Weitz, S.; Mwaikono, K.S.; Gardner-Lubbe, S.; Zar, H.J.; Nicol, M.P.; du Toit, E. The influence of DNA extraction and lipid removal on human milk bacterial profiles. Methods Protoc. 2020, 3, 39. [Google Scholar] [CrossRef]
- Cheema, A.S.; Stinson, L.F.; Lai, C.T.; Geddes, D.T.; Payne, M.S. DNA extraction method influences human milk bacterial profiles. J. Appl. Microbiol. 2021, 130, 142–156. [Google Scholar] [CrossRef]
- Stinson, L.F.; Trevenen, M.L.; Geddes, D.T. The viable microbiome of human milk differs from the metataxonomic profile. Nutrients 2021, 13, 4445. [Google Scholar] [CrossRef]
- Douglas, C.A.; Ivey, K.L.; Papanicolas, L.E.; Best, K.P.; Muhlhausler, B.S.; Rogers, G.B. DNA extraction approaches substantially influence the assessment of the human breast milk microbiome. Sci. Rep. 2020, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Siebert, A.; Hofmann, K.; Staib, L.; Doll, E.V.; Scherer, S.; Wenning, M. Amplicon-sequencing of raw milk microbiota: Impact of DNA extraction and library-PCR. Appl. Microbiol. Biotechnol. 2021, 105, 4761–4773. [Google Scholar] [CrossRef] [PubMed]
- William, S.; Feil, H.; Copeland, A. Bacterial DNA Isolation CTAB Protocol Bacterial Genomic DNA Isolation Using CTAB Materials & Reagents; Doe Joint Genome Institute: Berkeley, CA, USA, 2004; p. 4.
- Congreso de la Unión; Cámara de Diputados del H. De la Ley General de Salud. Diario Oficial de la Federación; Congreso de la Unión: Ciudad de México, México, 2018; pp. 1–342. [Google Scholar]
- Cámara de Diputados del H.; Congreso de la Union. Ley Federal de Protección de Datos Personales en Posesión de los Particulares; Congreso de la Union: Ciudad de México, México, 2010; pp. 1–18. [Google Scholar]
- Gámez-Valdez, J.S.; García-Mazcorro, J.F.; Montoya-Rincón, A.H.; Rodríguez-Reyes, D.L.; Jiménez-Blanco, G.; Rodríguez, M.T.A.; de Vaca, R.P.-C.; Alcorta-García, M.R.; Brunck, M.; Lara-Díaz, V.J.; et al. Differential analysis of the bacterial community in colostrum samples from women with gestational diabetes mellitus and obesity. Sci. Rep. 2021, 11, 24373. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and Direct Sequencing of Fungal Ribosomal Rna Genes for Phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar] [CrossRef]
- Lin, P. Genomic DNA QC Using Standard Gel Electrophoresis (for Collaborators); Doe Joint Genome Institute: Berkeley, CA, USA, 2012; pp. 1–11, Versión 3.
- Banco Nacional de ADN Carlos III. Programa de Control de Calidad de Ácidos Nucleicos; Universidad de Salamanca: Salamanca, Spain, 2020; pp. 1–10. [Google Scholar]
- Cohen, J. A ower primer. Psychol. Bull. 1992, 112, 155–159. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017; Volume 2, ISBN 3900051070. [Google Scholar]
- RStudio Team. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA, 2022. [Google Scholar]
- Moossavi, S.; Azad, M.B. Origins of human milk microbiota: New evidence and arising questions. Gut Microbes 2019, 12, 1667722. [Google Scholar] [CrossRef] [Green Version]
- Männistö, M.K.; Tiirola, M.; Häggblom, M.M. Effect of Freeze-Thaw Cycles on Bacterial Communities of Arctic Tundra Soil. Microb. Ecol. 2009, 58, 621–631. [Google Scholar] [CrossRef]
- Xin, Y.; Xie, J.; Nan, B.; Tang, C.; Xiao, Y.; Wu, Q.; Lin, Y.; Zhang, X.; Shen, H. Freeze-Thaw Pretreatment Can Improve Efficiency of Bacterial DNA Extraction From Meconium. Front. Microbiol. 2021, 12, 753688. [Google Scholar] [CrossRef]
- Rutherford, C.Z. Optimization of DNA Extraction from Human Milk. Master’s Thesis, Winthrop University, Rock Hill, SC, USA, 2020. [Google Scholar]
- Sambrook, J.; Russel, D.W. Molecular Cloning: A Laboratory Manual, 4th ed.; Cold Spring Harbor Lab Press: Cold Spring Harbor, NY, USA, 2012; Chapter 2; pp. 84–149. ISBN 9781936113415. [Google Scholar]
- Gaur, M.; Vasudeva, A.; Singh, A.; Sharma, V.; Khurana, H.; Negi, R.K.; Lee, J.-K.; Kalia, V.C.; Misra, R.; Singh, Y. Comparison of DNA Extraction Methods for Optimal Recovery of Metagenomic DNA from Human and Environmental Samples. Indian J. Microbiol. 2019, 59, 482–489. [Google Scholar] [CrossRef]
- Bermúdez, M.; Guzmán, S.; Orozco, M.; Velázquez, J.; Buenrostro, M.; Michel, C. Optimizing a protocol for DNA isolation of leaf Saccharum officinarum. Rev. Mex. Cienc. Agrícolas 2016, 7, 897–910. [Google Scholar] [CrossRef]
- Xue, Z.; Marco, M.L. Improved assessments of milk microbiota composition via sample preparation and DNA extraction methods. bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Nearing, J.T.; Comeau, A.M.; Langille, M.G.I. Identifying biases and their potential solutions in human microbiome studies. Microbiome 2021, 9, 113. [Google Scholar] [CrossRef] [PubMed]
- Safarikova, M.; Kubena, A.A.; Frankova, V.; Zima, T.; Kalousova, M. The effects of different storage conditions and repeated freeze/thaw cycles on the concentration, purity and integrity of genomic DNA. Folia Biol. 2021, 67, 10–15. [Google Scholar]
- Patil, A.; Dubey, A.; Malla, M.A.; Disouza, J.; Pawar, S.; Alqarawi, A.A.; Hashem, A.; Abd_Allah, E.F.; Kumar, A. Complete Genome Sequence of Lactobacillus plantarum Strain JDARSH, Isolated from Sheep Milk. Microbiol. Resour. Announc. 2020, 9, e01199-19. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, E.; Langa, S.; Martín, V.; Arroyo, R.; Martín, R.; Fernández, L.; Rodríguez, J.M. Complete genome sequence of Lactobacillus fermentum CECT 5716, a probiotic strain isolated from human milk. J. Bacteriol. 2010, 192, 4800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardy, E.E.; Petit, R.A.; Moller, A.G.; Blumenthal, J.A.; McAdam, A.J.; Priebe, G.P.; Chande, A.T.; Rishishwar, L.; Jordan, I.K.; Read, T.D.; et al. Whole-Genome Sequences of Staphylococcus aureus Isolates from Cystic Fibrosis Lung Infections. Microbiol. Resour. Announc. 2019, 8, 17–19. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, T.L.; Bayles, D.O.; Shore, S.M. Complete Genome Sequence of Bordetella bronchiseptica Strain KM22. Microbiol. Resour. Announc. 2020, 9, e01207-19. [Google Scholar] [CrossRef] [Green Version]
- Wan, K.H.; Park, S.; Hess, B.M.; Neff, M.J.; Booth, B.W.; Celniker, S.E. Complete Genome Sequence of the Citrobacter freundii Type Strain. Microbiol. Resour. Announc. 2020, 9, e00240-20. [Google Scholar] [CrossRef]
- Takeno, A.; Okamoto, A.; Tori, K.; Oshima, K.; Hirakawa, H.; Toh, H.; Agata, N.; Yamada, K.; Ogasawara, N.; Hayashi, T.; et al. Complete genome sequence of Bacillus cereus NC7401, which produces high levels of the emetic toxin cereulide. J. Bacteriol. 2012, 194, 4767–4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Zheng, B.; Ding, W.; Lv, L.; Ji, J.; Zhang, H.; Xiao, Y.; Li, L. Whole-Genome sequence of Staphylococcus hominis, an opportunistic pathogen. J. Bacteriol. 2012, 194, 4761–4762. [Google Scholar] [CrossRef] [Green Version]
- Leite, E.L.; Araújo, W.J.; Vieira, T.R.; Zenato, K.S.; Vasconcelos, P.C.; Cibulski, S.; Givisiez, P.E.N.; Cardoso, M.R.I.; Oliveira, C.J.B. First reported genome of an mcr-9-mediated colistin-resistant Salmonella Typhimurium isolate from Brazilian livestock. J. Glob. Antimicrob. Resist. 2020, 23, 394–397. [Google Scholar] [CrossRef]
- Clifford, R.J.; Hang, J.; Riley, M.C.; Onmus-Leone, F.; Kuschner, R.A.; Lesho, E.P.; Waterman, P.E. Complete genome sequence of Providencia stuartii clinical isolate MRSN 2154. J. Bacteriol. 2012, 194, 3736–3737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galac, M.R.; Stam, J.; Maybank, R.; Hinkle, M.; Mack, D.; Rohde, H.; Roth, A.L.; Fey, P.D. Complete genome sequence of Staphylococcus epidermidis 1457. Genome Announc. 2017, 5, e00450-17. [Google Scholar] [CrossRef] [Green Version]
- Kuo, V.; Shoemaker, W.R.; Muscarella, M.E.; Lennon, J.T. Whole-Genome Sequence of the Soil Bacterium Micrococcus sp. KBS0714. Genome Announc. 2013, 2662, e00697-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, C.; Nelson, M.; Cao, G.; Sadowsky, M.J.; Yan, T. crossm Complete Genome Sequence of the Disinfectant Susceptibility Testing Reference Strain Staphylococcus aureus subsp. aureus ATCC 6538. Genome Announc. 2017, 5, e00293-17. [Google Scholar]
- Mustafa, A.; Ibrahim, M.; Rasheed, M.A.; Kanwal, S.; Hussain, A.; Sami, A.; Ahmed, R.; Bo, Z. Genome-wide Analysis of Four Enterobacter cloacae complex type strains: Insights into Virulence and Niche Adaptation. Sci. Rep. 2020, 10, 8150. [Google Scholar] [CrossRef]
- Archer, C.T.; Kim, J.F.; Jeong, H.; Park, J.H.; Vickers, C.E.; Lee, S.Y.; Nielsen, L.K. The genome sequence of E. coli W (ATCC 9637): Comparative genome analysis and an improved genome-scale reconstruction of E. coli. BMC Genom. 2011, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Chénard, C.; Wong, A.; Kolundžija, S.; Clare, M.E.; Kushwaha, K.K.; Panicker, D.; Putra, A.; Gaultier, N.E.; Heinle, C.E.; Vettath, K.; et al. crossm Complete Genome Sequence of Staphylococcus haemolyticus. Genome Announc. 2018, 16, e00229-18. [Google Scholar]
- Minnullina, L.; Pudova, D.; Shagimardanova, E.; Shigapova, L.; Sharipova, M.; Mardanova, A. Comparative genome analysis of uropathogenic morganella morganii strains. Front. Cell. Infect. Microbiol. 2019, 9, 167. [Google Scholar] [CrossRef]
- Bitar, I.; Marchetti, V.M.; Mercato, A.; Nucleo, E.; Anesi, A.; Bracco, S.; Rognoni, V.; Hrabak, J.; Migliavacca, R. Complete genome and plasmids sequences of a clinical proteus mirabilis isolate producing plasmid mediated ndm-1 from Italy. Microorganisms 2020, 8, 339. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, Z.; Sam, N.; Vaidyanathan, R. Whole genome sequence of Klebsiella pneumoniae U25, a hypermucoviscous, multidrug resistant, biofilm producing isolate from India. Mem. Inst. Oswaldo Cruz 2016, 111, 144–146. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.R.; Park, G.S.; Asaf, S.; Hong, S.J.; Jung, B.K.; Shin, J.H. Complete genome analysis of Serratia marcescens RSC-14: A plant growth-promoting bacterium that alleviates cadmium stress in host plants. PLoS ONE 2017, 12, e0171534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, T.; Federspiel, N.A.; Chibana, H.; Dungan, J.; Kalman, S.; Magee, B.B.; Newport, G.; Thorstenson, Y.R.; Agabian, N.; Magee, P.T.; et al. The diploid genome sequence of Candida albicans. Proc. Natl. Acad. Sci. USA 2004, 101, 7329–7334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Yang, Y.; Zhao, T. Genome Sequence of Candida tropicalis no. 121, Used for RNA Production. Genome Announc. 2014, 3, e00316-14. [Google Scholar] [CrossRef]
- Gan, H.M.; Thomas, B.N.; Cavanaugh, N.T.; Morales, G.H.; Mayers, A.N.; Savka, M.A.; Hudson, A.O. Whole genome sequencing of Rhodotorula mucilaginosa isolated from the chewing stick (Distemonanthus benthamianus): Insights into Rhodotorula phylogeny, mitogenome dynamics and carotenoid biosynthesis. PeerJ 2017, 2017, e4030. [Google Scholar] [CrossRef] [Green Version]
- Alarcón-Zúñiga, B.; Zepeda-Batista, J.L.; Ruíz-Flores, A.; Gómez-Meza, L.J.; García-Muñiz, J.G.; Núñez-Domínguez, R.; Ramírez-Valverde, R.; Villegas-Velázquez, I. Modificación del método de tiocianato de guanidina para extraer ADN de semen para análisis genómico en mamíferos. Rev. Mex. Cienc. Pecu. 2016, 7, 405. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| DNA Extraction Method | ZYMO | GTC | CTAB-2PH | CTAB-STD | † p-Value | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Median | Mean ± SD | Median | Mean ± SD | Median | Mean ± SD | Median | ||
| DNA (ng/µL) | 5.39 ± 4.18 | 3.4 | 29.62 ± 18.07 | 36.85 f | 41.42 ± 12.93 | 31.65 e | 37.14 ± 22.57 | 31.19 c | 5.37 × 10−5 |
| Purity (260/280 nm) | 1.33 ± 0.08 | 1.3 | 1.1 ± 0.26 | 1.38 | 1.51 ± 0.09 | 1.51 e | 1.68 ± 0.12 | 1.67 a,b,c | 1.38 × 10−7 |
| Purity (260/230 nm) | 0.29 ± 0.14 f | 0.24 | 0.11 ± 0.08 | 0.1 | 0.56 ± 0.22 | 0.47 d | 0.58 ± 0.26 | 0.48 a,b | 1.09 × 10−9 |
| Integrity | 0.61 ± 0.8 | 0.25 | 2.68 ± 0.36 | 2.75 | 1.9 ± 0.58 | 2 d | 2.64 ± 0.54 | 3 a,c,e,f | 1.51 × 10−7 |
| Cell Type | DNA (ng/µL) | Purity (260/280 nm) | Purity (260/230 nm) | Integrity | ||||
|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Median | Mean ± SD | Median | Mean ± SD | Median | Mean ± SD | Median | |
| Gram(+) bacteria | 33.74 ± 25.83 a,b | 28.15 | 1.76 ± 0.14 | 1.83 | 1.35 ± 0.49 | 1.49 | 1.86 ± 0.69 | 2 |
| Gram(−) bacteria | 80.68 ± 32.89 c | 60.34 | 1.83 ± 0.09 | 1.86 | 1.6 ± 0.43 | 1.68 | 1.67 ± 0.5 | 2 |
| Yeast | 85.86 ± 34.84 | 97.22 | 1.75 ± 0.09 | 1.75 | 1.48 ± 0.14 | 1.48 | 2.25 ± 0.5 | 2 |
| p-value | 0.0133 † | 0.528 ¥ | 0.4454 ¥ | 0.2663 ¥ | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alemán-Duarte, M.I.; Aguilar-Uscanga, B.R.; García-Robles, G.; Ramírez-Salazar, F.d.J.; Benítez-García, I.; Balcázar-López, E.; Solís-Pacheco, J.R. Improvement and Validation of a Genomic DNA Extraction Method for Human Breastmilk. Methods Protoc. 2023, 6, 34. https://doi.org/10.3390/mps6020034
Alemán-Duarte MI, Aguilar-Uscanga BR, García-Robles G, Ramírez-Salazar FdJ, Benítez-García I, Balcázar-López E, Solís-Pacheco JR. Improvement and Validation of a Genomic DNA Extraction Method for Human Breastmilk. Methods and Protocols. 2023; 6(2):34. https://doi.org/10.3390/mps6020034
Chicago/Turabian StyleAlemán-Duarte, Mario Iván, Blanca Rosa Aguilar-Uscanga, Guadalupe García-Robles, Felipe de Jesús Ramírez-Salazar, Israel Benítez-García, Edgar Balcázar-López, and Josué Raymundo Solís-Pacheco. 2023. "Improvement and Validation of a Genomic DNA Extraction Method for Human Breastmilk" Methods and Protocols 6, no. 2: 34. https://doi.org/10.3390/mps6020034