
Identification and Characterisation of Infiltrating Immune Cells in Malignant Pleural Mesothelioma Using Spatial Transcriptomics

 , , , , , , , , and
, , , , , , , , and
Abstract
:1. Background
2. Experimental Design
2.1. Tissue Samples
2.2. Purchased Consumables
- Visium Spatial for FFPE Gene Expression kit, Human Transcriptome (10x Genomics, Pleasanton, CA, USA, 1000338);
- 10x Visium Accessory kit (10x Genomics, 1000194);
- Visium Tissue Section test slides (10x Genomics, 1000347);
- Dual Index kit TS Set A, 96 runs (10x Genomics, 1000251);
- RNeasy FFPE kit (Qiagen, Hilden, Germany, 73504);
- Deparaffinisation solution (Qiagen, 19093);
- Tween-20 (Sigma-Aldrich, St. Louis, MO, USA 9005-64-5);
- 10x phosphate-buffered saline pH 7.4 (ThermoFisher Scientific, Waltham, MA, USA, 70011609);
- 20x SSC buffer (ThermoFisher Scientific, 15557044);
- 8 M KOH (Merck, Darmstadt, Germany, 1310-58-3);
- 1 M Tris, pH 7.0, RNase free (ThermoFisher Scientific, AM9851);
- Low-Profile/triple facet microtome blades (Leica Biosystems, Wetzlar, Germany, 3802112);
- Kapa SYBR Fast qPCR Master Mix (2x), 1 mL (Sigma-Aldrich, KK4602);
- SPRIselect reagent, 450 mL (Beckman Coulter, Brea, CA, USA, B23319).
2.3. Equipment
- RM2125RT rotary microtome (Leica Biosystems);
- VS120 Slide Scanner (Olympus, Tokyo, Japan);
- Veriti 96-well thermocycler (Applied Biosystems, Waltham, MA, USA);
- 4200 TapeStation System (Agilent Technologies, Santa Clara, CA, USA).
2.4. Other Services
- Library quality control (QC) at AGRF (Australian Genome Research Facility, Melbourne, VIC, Australia);
- Sequencing at AGRF—Illumina Novaseq 6000 SP reagent kit v1.5, 50 bp PE.
3. Procedure
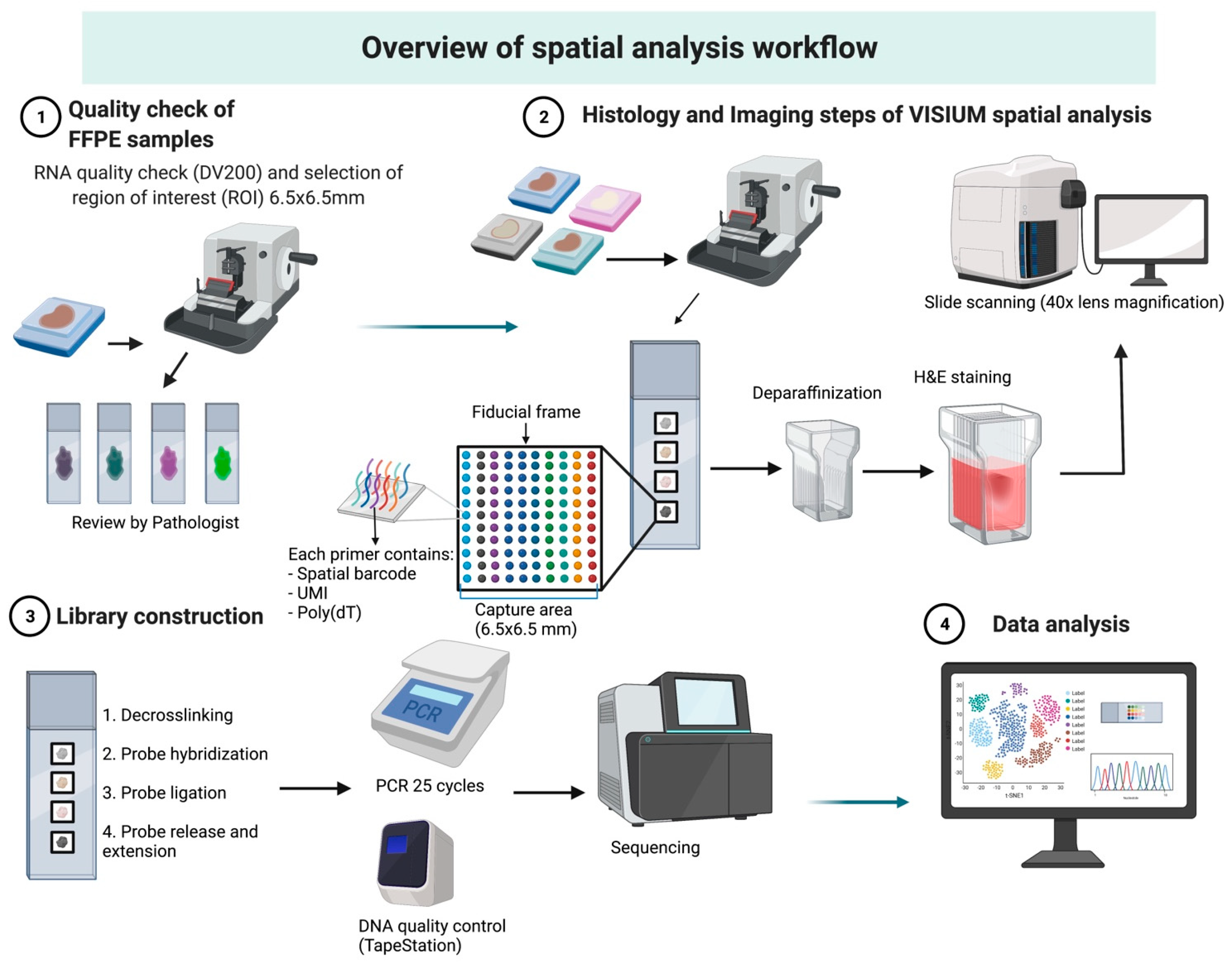
3.1. Overview of Workflow
3.1.1. Case Selection and Tissue Preparation
3.1.2. RNA Quality Check
3.1.3. Visium Tissue Section Test Slide
3.2. Histology and Imaging
3.3. Library Preparation and Sequencing
3.4. Bioinformatic Analysis
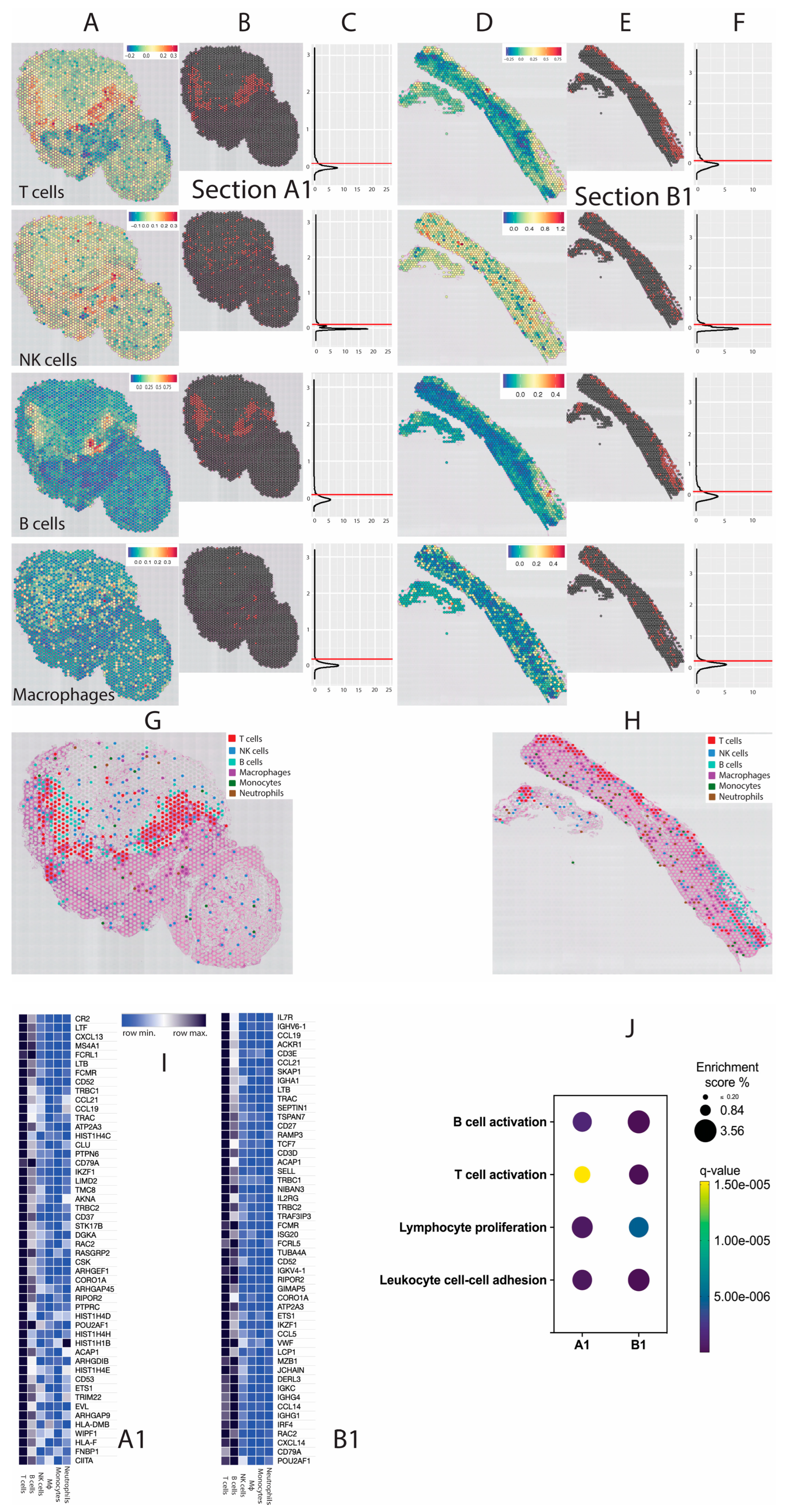
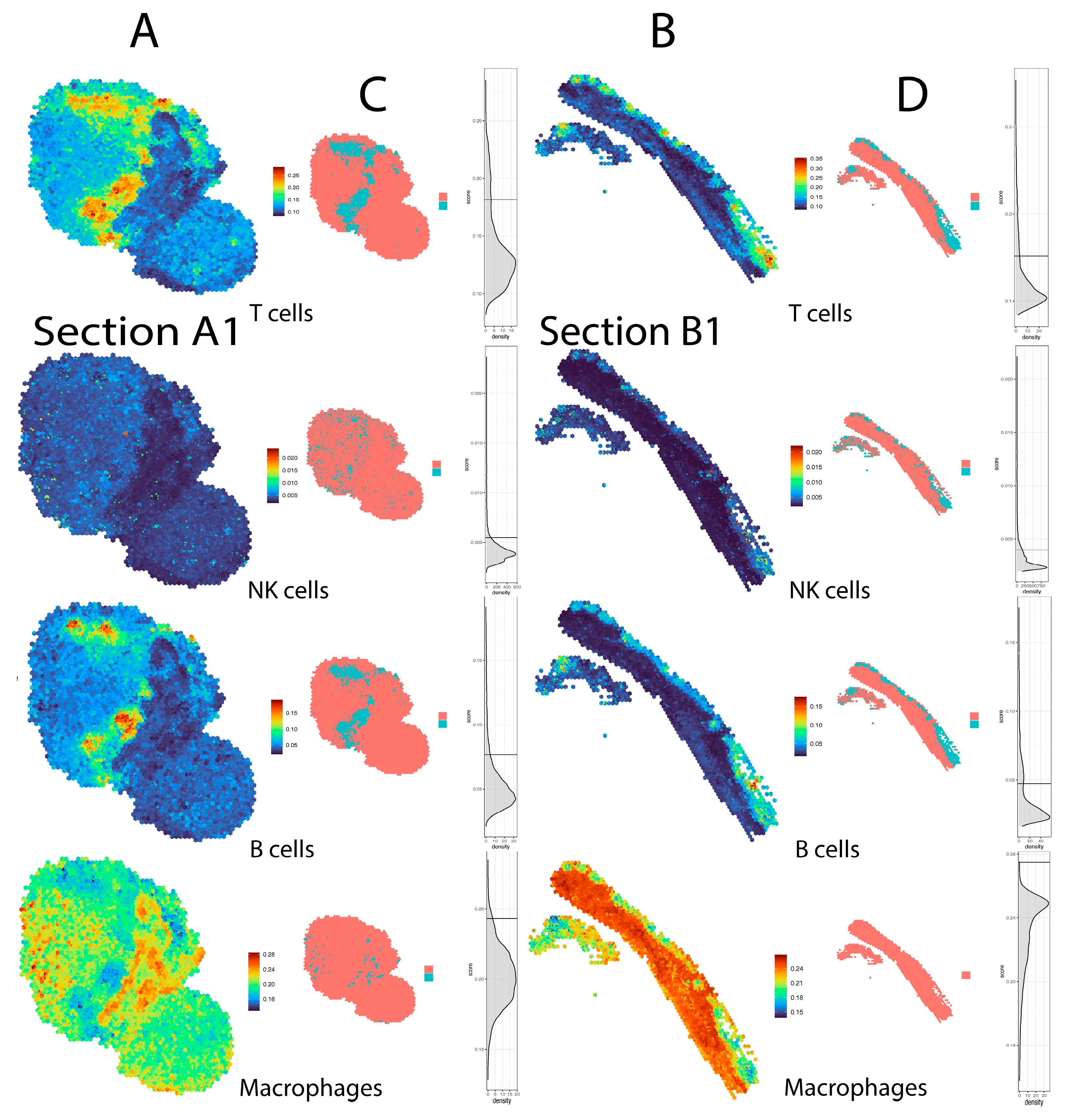
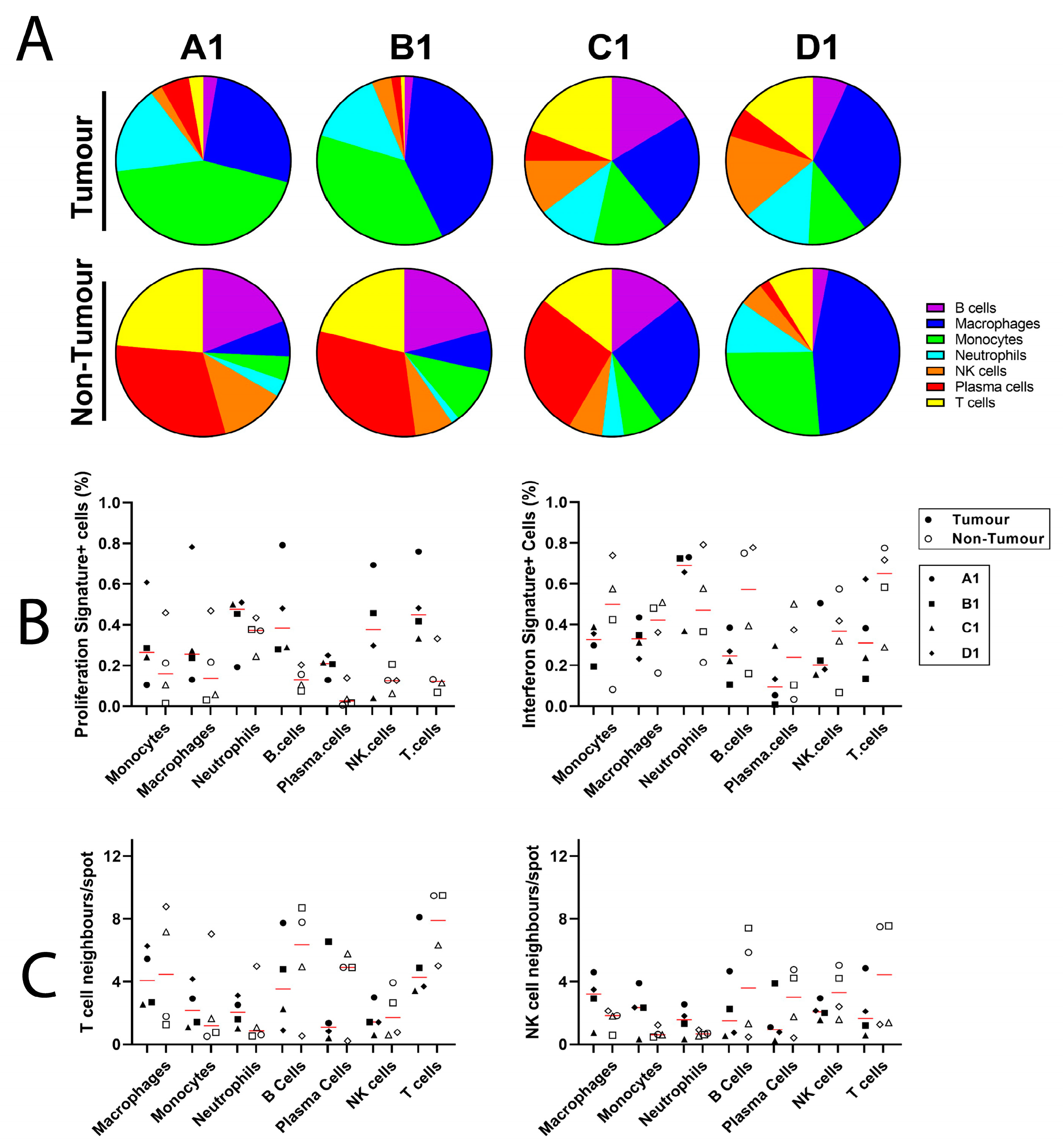
4. Results
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Popat, S.; Baas, P.; Faivre-Finn, C.; Girard, N.; Nicholson, A.G.; Nowak, A.K.; Opitz, I.; Scherpereel, A.; Reck, M. Malignant pleural mesothelioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2022, 33, 129–142. [Google Scholar] [CrossRef]
- Alpert, N.; van Gerwen, M.; Taioli, E. Epidemiology of mesothelioma in the 21(st) century in Europe and the United States, 40 years after restricted/banned asbestos use. Transl. Lung Cancer Res. 2020, 9 (Suppl. S1), S28–S38. [Google Scholar] [CrossRef] [PubMed]
- Eberst, G.; Anota, A.; Scherpereel, A.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Lena, H.; et al. Health-Related Quality of Life Impact from Adding Bevacizumab to Cisplatin-Pemetrexed in Malignant Pleural Mesothelioma in the MAPS IFCT-GFPC-0701 Phase III Trial. Clin. Cancer Res. 2019, 25, 5759–5765. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): A multicentre, randomised, open-label, phase 3 trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Shek, D.; Akhuba, L.; Carlino, M.S.; Nagrial, A.; Moujaber, T.; Read, S.A.; Gao, B.; Ahlenstiel, G. Immune-Checkpoint Inhibitors for Metastatic Colorectal Cancer: A Systematic Review of Clinical Outcomes. Cancers 2021, 13, 4345. [Google Scholar] [CrossRef]
- Shek, D.; Read, S.A.; Nagrial, A.; Carlino, M.S.; Gao, B.; George, J.; Ahlenstiel, G. Immune-Checkpoint Inhibitors for Advanced Hepatocellular Carcinoma: A Synopsis of Response Rates. Oncologist 2021, 26, e1216–e1225. [Google Scholar] [CrossRef]
- Bertrand, A.; Kostine, M.; Barnetche, T.; Truchetet, M.E.; Schaeverbeke, T. Immune related adverse events associated with anti-CTLA-4 antibodies: Systematic review and meta-analysis. BMC Med. 2015, 13, 211. [Google Scholar] [CrossRef] [Green Version]
- Maughan, B.L.; Bailey, E.; Gill, D.M.; Agarwal, N. Incidence of Immune-Related Adverse Events with Program Death Receptor-1- and Program Death Receptor-1 Ligand-Directed Therapies in Genitourinary Cancers. Front. Oncol. 2017, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Puzanov, I.; Diab, A.; Abdallah, K.; Bingham, C.O., 3rd; Brogdon, C.; Dadu, R.; Hamad, L.; Kim, S.; Lacouture, M.E.; LeBoeuf, N.R.; et al. Managing toxicities associated with immune checkpoint inhibitors: Consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J. Immunother. Cancer 2017, 5, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haanen, J.; Ernstoff, M.S.; Wang, Y.; Menzies, A.M.; Puzanov, I.; Grivas, P.; Larkin, J.; Peters, S.; Thompson, J.A.; Obeid, M. Autoimmune diseases and immune-checkpoint inhibitors for cancer therapy: Review of the literature and personalized risk-based prevention strategy. Ann. Oncol. 2020, 31, 724–744. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Huang, X.; Zhang, G.; Hong, Z.; Bai, X.; Liang, T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal. Transduct. Target Ther. 2021, 6, 72. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e3529. [Google Scholar] [CrossRef]
- Zhao, E.; Stone, M.R.; Ren, X.; Guenthoer, J.; Smythe, K.S.; Pulliam, T.; Williams, S.R.; Uytingco, C.R.; Taylor, S.E.B.; Nghiem, P.; et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 2021, 39, 1375–1384. [Google Scholar] [CrossRef]
- Nirmal, A.J.; Regan, T.; Shih, B.B.; Hume, D.A.; Sims, A.H.; Freeman, T.C. Immune Cell Gene Signatures for Profiling the Microenvironment of Solid Tumors. Cancer Immunol. Res. 2018, 6, 1388–1400. [Google Scholar] [CrossRef] [Green Version]
- Barone, E.; Gemignani, F.; Landi, S. Overexpressed genes in malignant pleural mesothelioma: Implications in clinical management. J. Thorac. Dis. 2018, 10 (Suppl. S2), S369–S382. [Google Scholar] [CrossRef] [Green Version]
- Valdes-Mora, F.; Salomon, R.; Gloss, B.S.; Law, A.M.K.; Venhuizen, J.; Castillo, L.; Murphy, K.J.; Magenau, A.; Papanicolaou, M.; Rodriguez de la Fuente, L.; et al. Single-cell transcriptomics reveals involution mimicry during the specification of the basal breast cancer subtype. Cell Rep. 2021, 35, 108945. [Google Scholar] [CrossRef]
- Pearson, R.K.; Neuvo, Y.; Astola, J.; Gabbouj, M. Generalized Hampel Filters. EURASIP J. Adv. Signal Process. 2016, 2016, 87. [Google Scholar] [CrossRef] [Green Version]
- Method of the Year 2020: Spatially resolved transcriptomics. Nat Methods 2021, 18, 1. [CrossRef]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Alay, A.; Cordero, D.; Hijazo-Pechero, S.; Aliagas, E.; Lopez-Doriga, A.; Marin, R.; Palmero, R.; Llatjos, R.; Escobar, I.; Ramos, R.; et al. Integrative transcriptome analysis of malignant pleural mesothelioma reveals a clinically relevant immune-based classification. J. Immunother. Cancer 2021, 9, e001601. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Bergenstrahle, J.; Lundeberg, J. Spatially Resolved Transcriptomes-Next Generation Tools for Tissue Exploration. Bioessays 2020, 42, e1900221. [Google Scholar] [CrossRef]
- Moses, L.; Pachter, L. Museum of spatial transcriptomics. Nat. Methods 2022, 19, 534–546. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Sub-Steps | Location | Estimated Timing |
|---|---|---|---|
| Case selection and quality control |
|
|
|
| FFPE Tissue Sectioning and Placement on Visium Slide |
|
|
|
| Deparaffinisation, H&E Staining, Imaging, Decrosslinking |
|
|
|
| Probe hybridisation |
|
|
|
| FFPE Library construction |
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shek, D.; Gloss, B.; Lai, J.; Ma, L.; Zhang, H.E.; Carlino, M.S.; Mahajan, H.; Nagrial, A.; Gao, B.; Read, S.A.; et al. Identification and Characterisation of Infiltrating Immune Cells in Malignant Pleural Mesothelioma Using Spatial Transcriptomics. Methods Protoc. 2023, 6, 35. https://doi.org/10.3390/mps6020035
Shek D, Gloss B, Lai J, Ma L, Zhang HE, Carlino MS, Mahajan H, Nagrial A, Gao B, Read SA, et al. Identification and Characterisation of Infiltrating Immune Cells in Malignant Pleural Mesothelioma Using Spatial Transcriptomics. Methods and Protocols. 2023; 6(2):35. https://doi.org/10.3390/mps6020035
Chicago/Turabian StyleShek, Dmitrii, Brian Gloss, Joey Lai, Li Ma, Hui E. Zhang, Matteo S. Carlino, Hema Mahajan, Adnan Nagrial, Bo Gao, Scott A. Read, and et al. 2023. "Identification and Characterisation of Infiltrating Immune Cells in Malignant Pleural Mesothelioma Using Spatial Transcriptomics" Methods and Protocols 6, no. 2: 35. https://doi.org/10.3390/mps6020035