
Principal Component Analysis versus Subject’s Residual Profile Analysis for Neuroinflammation Investigation in Parkinson Patients: A PET Brain Imaging Study


Abstract
:1. Introduction
2. Material and Methods
2.1. Subject and Data Acquisition
2.2. Image Registration
2.3. Demographic and Clinical Characteristics of Subjects
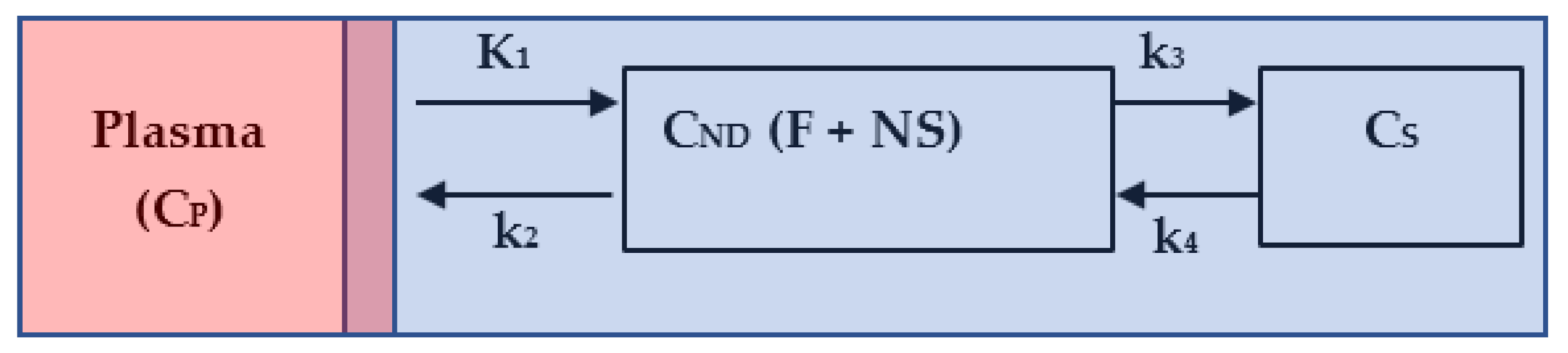
2.4. Kinetic Modeling
2.5. 11C-PBR28 Image Analysis
2.6. Statistical Analyses
2.7. Principal Component Analysis Definition and Process
3. Results
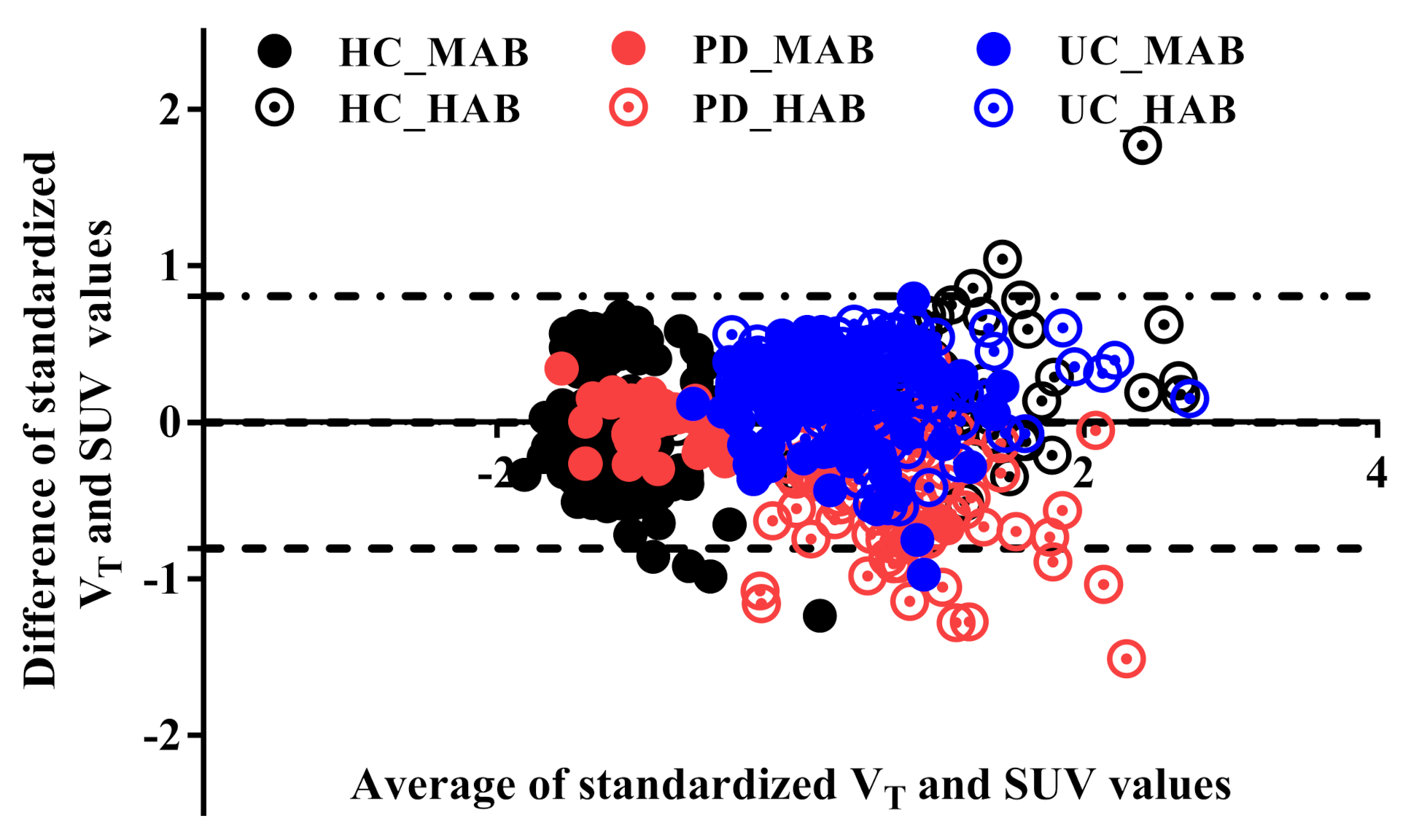
3.1. Validation of Standard Uptake Value measurement
3.2. Standard Uptake Value Profile Analysis
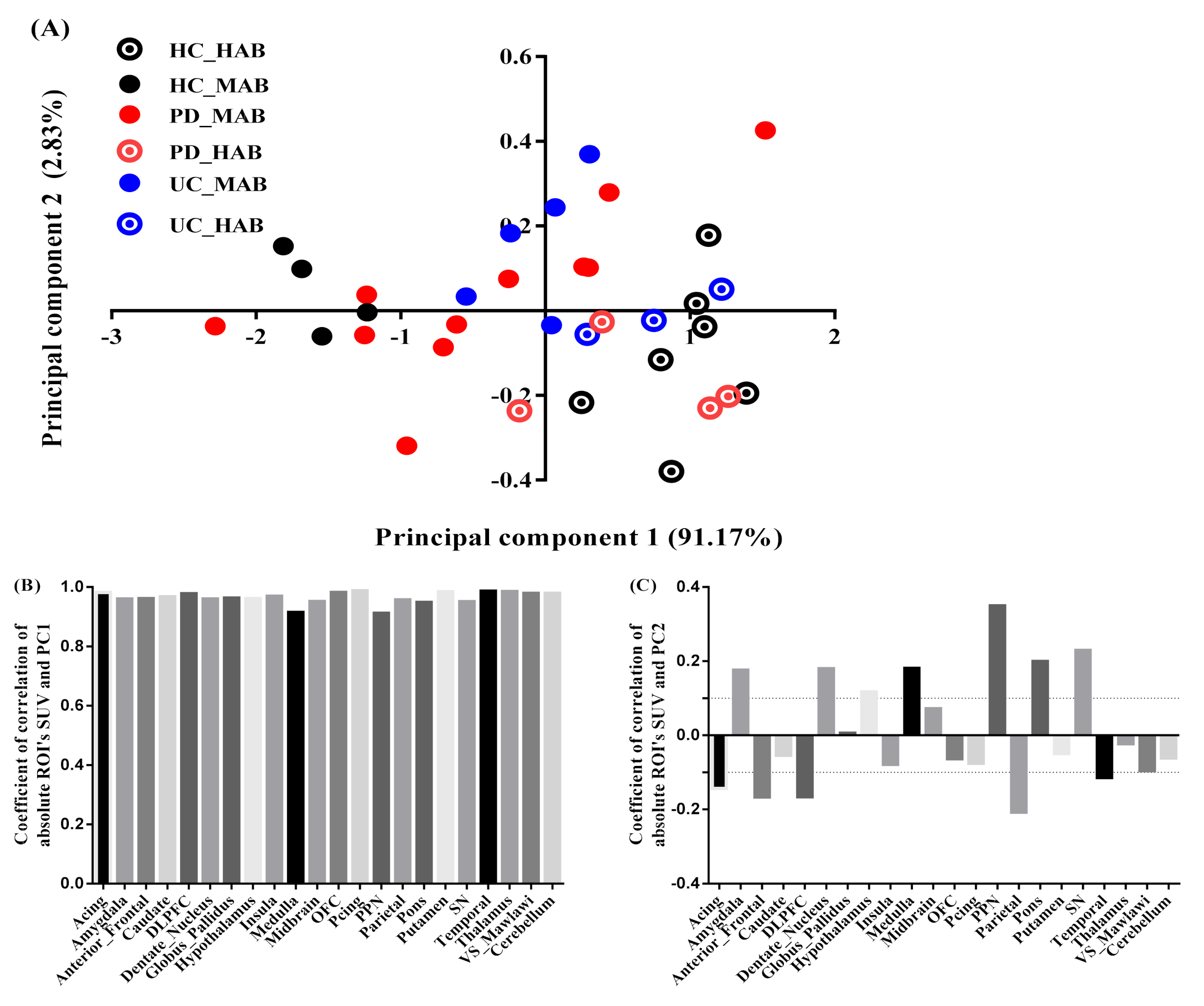
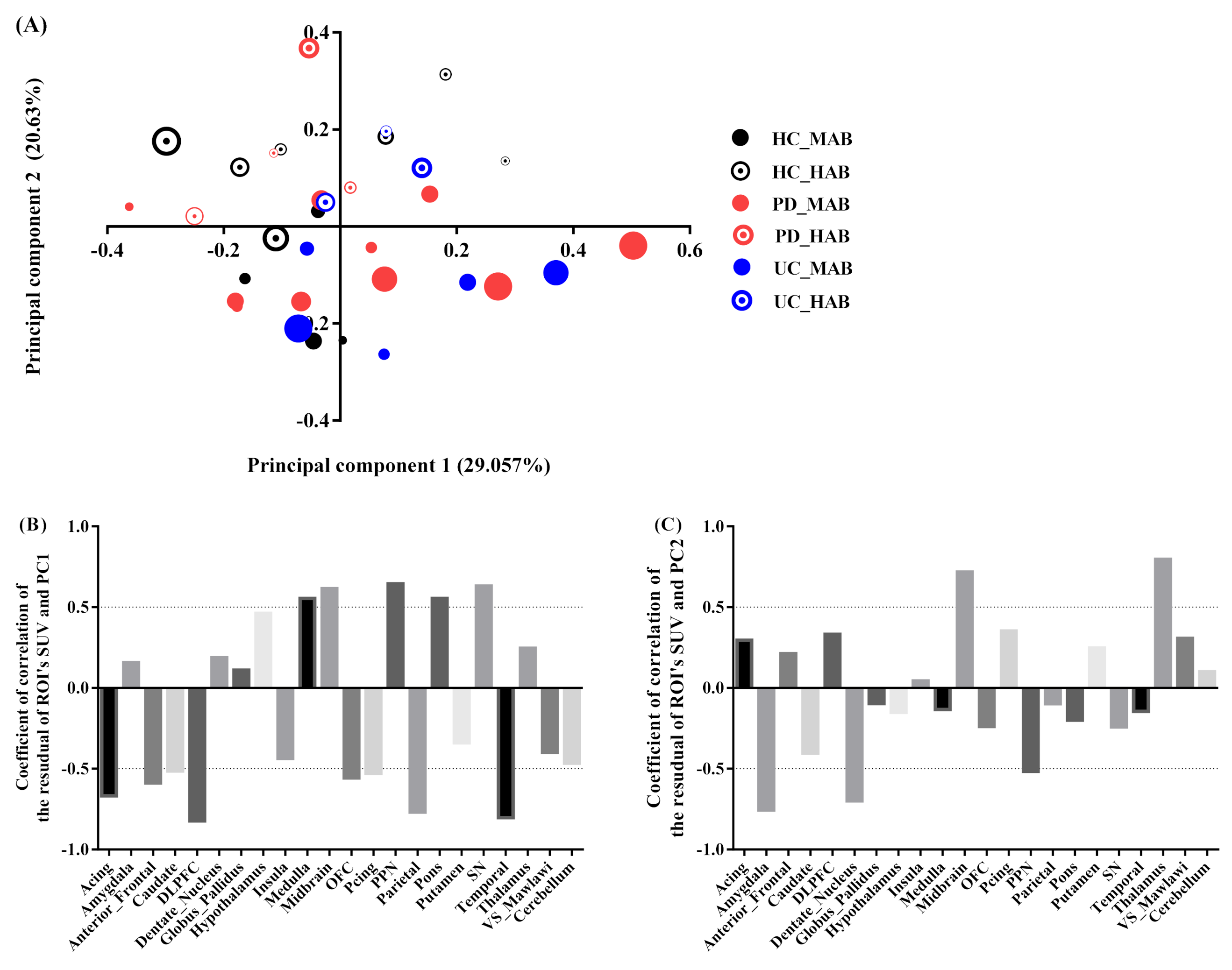
3.3. Principal Component Analysis
- (1)
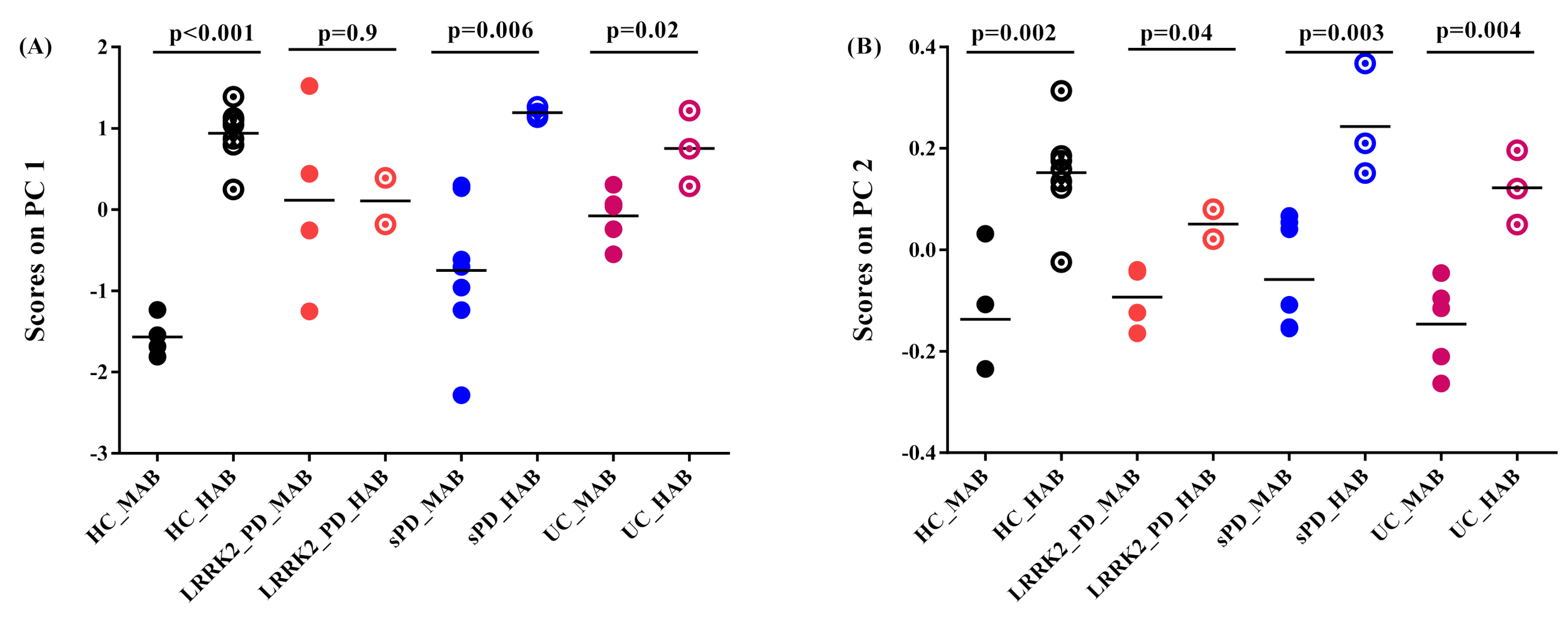
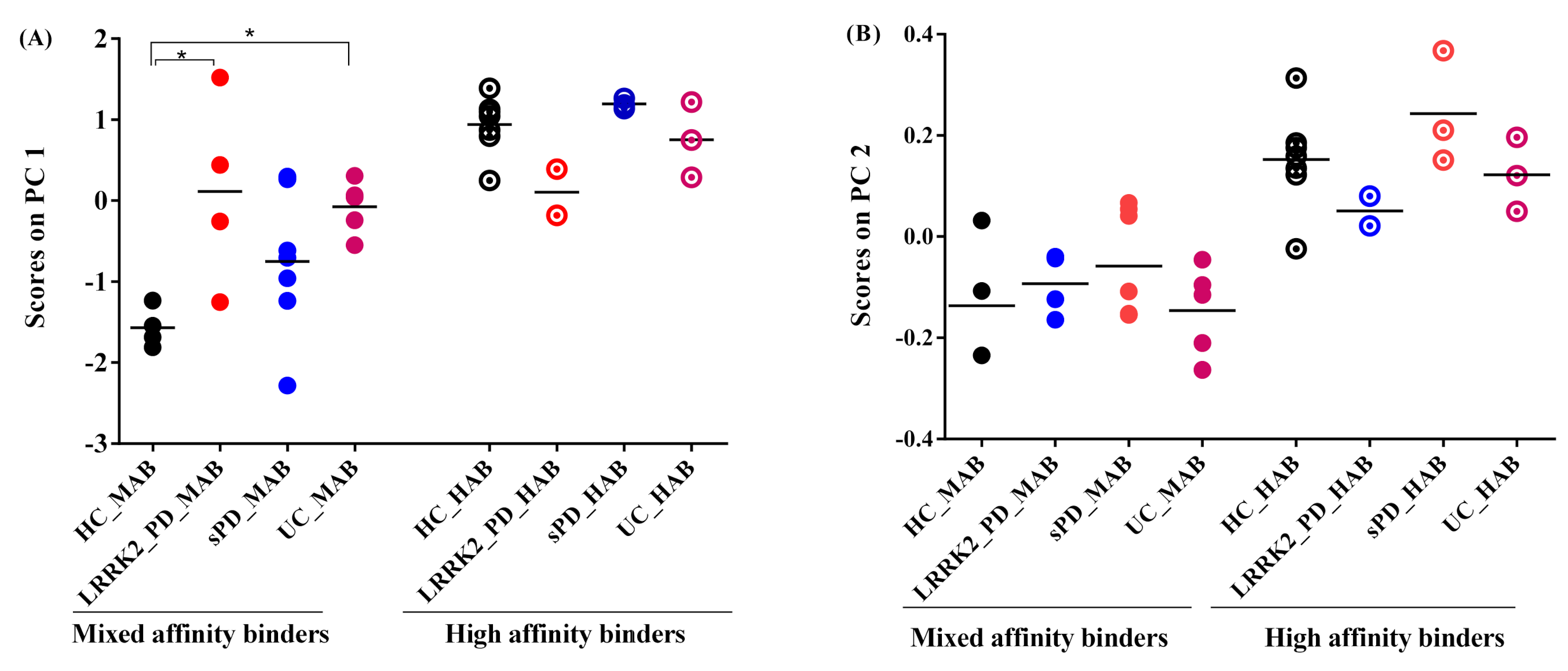
- Projection of absolute SUV on principal components
- (2)
- Projection of the subject’s residual profile on principal components
- (3)
- Effect of genotype on 11C-PBR28 SUV
- (4)
- Effect of disease on 11C-PBR28 SUV
- (5)
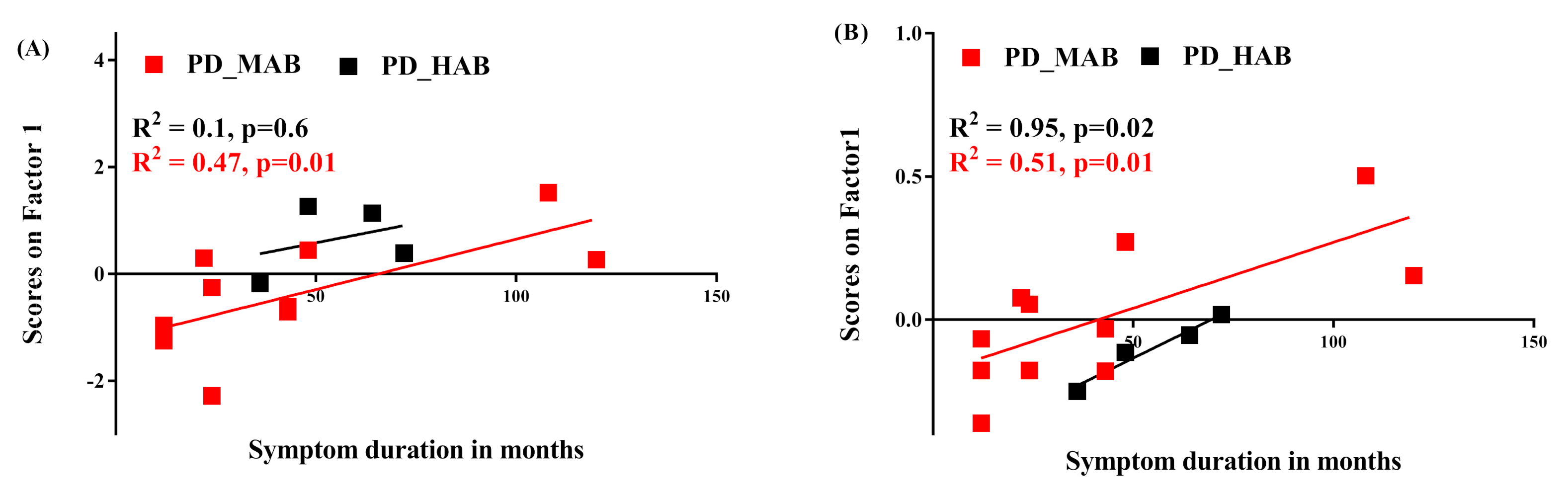
- Correlation between 11C-PBR28 SUV and symptom duration
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janssen, B.; Vugts, D.J.; Funke, U.; Molenaar, G.T.; Kruijer, P.S.; van Berckel, B.N.; Lammertsma, A.A.; Windhorst, A.D. Imaging of neuroinflammation in Alzheimer’s disease, multiple sclerosis and stroke: Recent developments in positron emission tomography. Biochim. Biophys. Acta 2016, 1862, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.D. Neuroinflammation. Neurosci. Bull. 2015, 31, 714–716. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Okello, A.A.; Brooks, D.J.; Edison, P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain 2015, 138 Pt 12, 3685–3698. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, O.; Kubicki, M.; Shenton, M.E. In vivo imaging of neuroinflammation in schizophrenia. Schizophr. Res. 2016, 173, 200–212. [Google Scholar] [CrossRef]
- Niccolini, F.; Su, P.; Politis, M. PET in multiple sclerosis. Clin. Nucl. Med. 2015, 40, e46–e52. [Google Scholar] [CrossRef]
- Banati, R.B.; Newcombe, J.; Gunn, R.N.; Cagnin, A.; Turkheimer, F.; Heppner, F.; Price, G.; Wegner, F.; Giovannoni, G.; Miller, D.H.; et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: Quantitative in vivo imaging of microglia as a measure of disease activity. Brain 2000, 123 Pt 11, 2321–2337. [Google Scholar] [CrossRef]
- Streit, W.J.; Xue, Q.S. Microglia in dementia with Lewy bodies. Brain Behav. Immun. 2016, 55, 191–201. [Google Scholar] [CrossRef]
- Surendranathan, A.; Rowe, J.B.; O’Brien, J.T. Neuroinflammation in Lewy body dementia. Parkinsonism Relat. Disord. 2015, 21, 1398–1406. [Google Scholar] [CrossRef]
- Koshimori, Y.; Ko, J.H.; Mizrahi, R.; Rusjan, P.; Mabrouk, R.; Jacobs, M.F.; Christopher, L.; Hamani, C.; Lang, A.E.; Wilson, A.A.; et al. Imaging Striatal Microglial Activation in Patients with Parkinson’s Disease. PLoS ONE 2015, 10, e0138721. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Kalogiannis, M.; Krasnikov, B.F.; Gomlin, I.; Peltier, M.R.; Moran, G.R. Linking Inflammation and Parkinson Disease: Hypochlorous Acid Generates Parkinsonian Poisons. Toxicol. Sci. 2016, 151, 388–402. [Google Scholar] [CrossRef] [PubMed]
- Rocha, N.P.; de Miranda, A.S.; Teixeira, A.L. Insights into Neuroinflammation in Parkinson’s Disease: From Biomarkers to Anti-Inflammatory Based Therapies. BioMed Res. Int. 2015, 2015, 628192. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, Y.; Yagi, S.; Yokokura, M.; Sakamoto, M. Neuroinflammation in the living brain of Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15 (Suppl. 3), S200–S204. [Google Scholar] [CrossRef]
- Machado, A.; Herrera, A.J.; Venero, J.L.; Santiago, M.; de Pablos, R.M.; Villaran, R.F.; Espinosa-Oliva, A.M.; Arguelles, S.; Sarmiento, M.; Delgado-Cortes, M.J.; et al. Inflammatory Animal Model for Parkinson’s Disease: The Intranigral Injection of LPS Induced the Inflammatory Process along with the Selective Degeneration of Nigrostriatal Dopaminergic Neurons. ISRN Neurol. 2011, 2011, 476158. [Google Scholar] [CrossRef]
- Liu, M.; Bing, G. Lipopolysaccharide animal models for Parkinson’s disease. Parkinsons Dis. 2011, 2011, 327089. [Google Scholar] [CrossRef]
- De Rosa, A.; De Michele, G.; Guacci, A.; Carbone, R.; Lieto, M.; Peluso, S.; Picillo, M.; Barone, P.; Salemi, F.; Laiso, A.; et al. Genetic screening for the LRRK2 R1441C and G2019S mutations in Parkinsonian patients from Campania. J. Parkinsons Dis. 2014, 4, 123–128. [Google Scholar] [CrossRef]
- De Rosa, A.; Criscuolo, C.; Mancini, P.; De Martino, M.; Giordano, I.A.; Pappata, S.; Filla, A.; De Michele, G. Genetic screening for LRRK2 gene G2019S mutation in Parkinson’s disease patients from Southern Italy. Parkinsonism Relat. Disord. 2009, 15, 242–244. [Google Scholar] [CrossRef]
- Deng, H.; Le, W.; Guo, Y.; Hunter, C.B.; Xie, W.; Huang, M.; Jankovic, J. Genetic analysis of LRRK2 mutations in patients with Parkinson disease. J. Neurol. Sci. 2006, 251, 102–106. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Abdelmotilib, H.; Liu, Z.; Stoyka, L.; Daher, J.P.; Milnerwood, A.J.; Unni, V.K.; Hirst, W.D.; Yue, Z.; Zhao, H.T.; et al. G2019S-LRRK2 Expression Augments alpha-Synuclein Sequestration into Inclusions in Neurons. J. Neurosci. 2016, 36, 7415–7427. [Google Scholar] [CrossRef] [PubMed]
- Gillardon, F.; Schmid, R.; Draheim, H. Parkinson’s disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience 2012, 208, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Bubacco, L.; Greggio, E. LRRK2 and neuroinflammation: Partners in crime in Parkinson’s disease? J. Neuroinflamm. 2014, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. LRRK2 Pathways Leading to Neurodegeneration. Curr. Neurol. Neurosci. Rep. 2015, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Cookson, M.R. Role of LRRK2 kinase dysfunction in Parkinson disease. Expert. Rev. Mol. Med. 2011, 13, e20. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Q.; Tan, L.; Yu, J.T. The role of the LRRK2 gene in Parkinsonism. Mol. Neurodegener. 2014, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Kim, J.W.; Dawson, V.L.; Dawson, T.M. LRRK2 pathobiology in Parkinson’s disease—Virtual inclusion. J. Neurochem. 2016, 139, 75–76. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Apel, A.; Schulte, C.; Schneiderhan-Marra, N.; Pont-Sunyer, C.; Vilas, D.; Ruiz-Martinez, J.; Langkamp, M.; Corvol, J.C.; Cormier, F.; et al. Inflammatory profile in LRRK2-associated prodromal and clinical PD. J. Neuroinflamm. 2016, 13, 122. [Google Scholar] [CrossRef]
- Lavisse, S.; Garcia-Lorenzo, D.; Peyronneau, M.A.; Bodini, B.; Thiriez, C.; Kuhnast, B.; Comtat, C.; Remy, P.; Stankoff, B.; Bottlaender, M. Optimized Quantification of Translocator Protein Radioligand (1)(8)F-DPA-714 Uptake in the Brain of Genotyped Healthy Volunteers. J. Nucl. Med. 2015, 56, 1048–1054. [Google Scholar] [CrossRef]
- Owen, D.R.; Yeo, A.J.; Gunn, R.N.; Song, K.; Wadsworth, G.; Lewis, A.; Rhodes, C.; Pulford, D.J.; Bennacef, I.; Parker, C.A.; et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. 2012, 32, 1–5. [Google Scholar] [CrossRef]
- Mizrahi, R.; Rusjan, P.M.; Kennedy, J.; Pollock, B.; Mulsant, B.; Suridjan, I.; De Luca, V.; Wilson, A.A.; Houle, S. Translocator protein (18 kDa) polymorphism (rs6971) explains in-vivo brain binding affinity of the PET radioligand [(18)F]-FEPPA. J. Cereb. Blood Flow Metab. 2012, 32, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Lyoo, C.H.; Ikawa, M.; Liow, J.S.; Zoghbi, S.S.; Morse, C.L.; Pike, V.W.; Fujita, M.; Innis, R.B.; Kreisl, W.C. Cerebellum Can Serve As a Pseudo-Reference Region in Alzheimer Disease to Detect Neuroinflammation Measured with PET Radioligand Binding to Translocator Protein. J. Nucl. Med. 2015, 56, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Oh, U.; Fujita, M.; Ikonomidou, V.N.; Evangelou, I.E.; Matsuura, E.; Harberts, E.; Fujimura, Y.; Richert, N.D.; Ohayon, J.; Pike, V.W.; et al. Translocator protein PET imaging for glial activation in multiple sclerosis. J. Neuroimmune Pharmacol. 2011, 6, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, P.S.; Selvaraj, S.; Veronese, M.; Rizzo, G.; Bertoldo, A.; Owen, D.R.; Bloomfield, M.A.; Bonoldi, I.; Kalk, N.; Turkheimer, F.; et al. Microglial Activity in People at Ultra High Risk of Psychosis and in Schizophrenia: An [(11)C]PBR28 PET Brain Imaging Study. Am. J. Psychiatry 2016, 173, 44–52. [Google Scholar] [CrossRef]
- Comtat, C.; Bataille, F.; Michel, C.; Jones, J.P.; Sibomana, M.; Janeiro, L.; Trebossen, R. OSEM-3D reconstruction strategies for the ECAT HRRT. In Proceedings of the IEEE Symposium Conference Record Nuclear Science 2004, Rome, Italy, 16–22 October 2004; Volume 3496, pp. 3492–3496. [Google Scholar]
- Logan, J.; Fowler, J.S.; Volkow, N.D.; Wolf, A.P.; Dewey, S.L.; Schlyer, D.J.; MacGregor, R.R.; Hitzemann, R.; Bendriem, B.; Gatley, S.J.; et al. Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl]-(-)-cocaine PET studies in human subjects. J. Cereb. Blood Flow Metab. 1990, 10, 740–747. [Google Scholar] [CrossRef]
- Ichise, M.; Toyama, H.; Innis, R.B.; Carson, R.E. Strategies to improve neuroreceptor parameter estimation by linear regression analysis. J. Cereb. Blood Flow Metab. 2002, 22, 1271–1281. [Google Scholar] [CrossRef]
- Laruelle, M.; Slifstein, M.; Huang, Y. Positron emission tomography: Imaging and quantification of neurotransporter availability. Methods 2002, 27, 287–299. [Google Scholar] [CrossRef]
- Abdi, H. Principal component analysis. Wiley Interdiscip. Rev. Comput. Stat. 2010, 2, 26. [Google Scholar] [CrossRef]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Bartels, A.L.; Willemsen, A.T.M.; Doorduin, J.; de Vries, E.F.J.; Dierckx, R.A.; Leenders, K.L. [11C]-PK11195 PET: Quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat. Disord. 2010, 16, 57–59. [Google Scholar] [CrossRef]
- Terada, T.; Yokokura, M.; Yoshikawa, E.; Futatsubashi, M.; Kono, S.; Konishi, T.; Miyajima, H.; Hashizume, T.; Ouchi, Y. Extrastriatal spreading of microglial activation in Parkinson’s disease: A positron emission tomography study. Ann. Nucl. Med. 2016, 30, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, C.; Tramontano, L.; Fiorenza, D.; Alfano, V.; Aiello, M.; Salvatore, M. Gliosis and neurodegenerative diseases: The role of PET and MR imaging. Front. Cell. Neurosci. 2020, 14, 75. [Google Scholar] [CrossRef] [PubMed]
- Van Weehaeghe, D.; Koole, M.; Schmidt, M.E.; Deman, S.; Jacobs, A.H.; Souche, E.; Serdons, K.; Sunaert, S.; Bormans, G.; Vandenberghe, W.; et al. [11C]JNJ54173717, a novel P2X7 receptor radioligand as marker for neuroinflammation: Human biodistribution, dosimetry, brain kinetic modelling and quantification of brain P2X7 receptors in patients with Parkinson’s disease and healthy volunteers. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2051–2064. [Google Scholar] [CrossRef] [PubMed]
- Rundo, L.; Militello, C.; Vitabile, S.; Russo, G.; Sala, E.; Gilardi, M.C. A Survey on Nature-Inspired Medical Image Analysis: A Step Further in Biomedical Data Integration. Fundam. Inform. 2020, 171, 345–365. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subjects | Mixed Affinity Binders (MAB) | High-Affinity Binders (MAB) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HC | sPD | Lrrk2-PD | lrrk2-UC | HC | sPD | Lrrk2-PD | lrrk2-UC | ||
| Age | 44.2 ± 16 | 6 ± 12 | 67 ± 17 | 51 ± 11 | 62 ± 16 | 56 ± 7 | 56 ± 3 | 53 ± 4 | |
| Gender | Male Female | 4 0 | 4 3 | 3 1 | 3 2 | 3 4 | 3 0 | 0 2 | 1 2 |
| UPDRS | 3 ± 2 | 23 ± 8 | 24 ± 6 | 1 ± 2 | 4 ± 1 | 24 ± 5 | 25 ± 7 | 0.6 ± 1 | |
| DD | NA | 3 ± 3 | 4 ± 3 | NA | NA | 4 ± 0.5 | 4.5 ± 2 | NA | |
| Tracer dose parameters | AI SA IM | 743 ± 0.7 93 ± 53 3.62 ± 0.9 | 623 ± 76 179 ± 128 1.22 ± 0.5 | 634 ± 82 172 ± 111 1.32 ± 0.8 | 696 ± 104 158 ± 125 1.3 ± 0.6 | 667 ± 80 215 ± 130 1.25 ± 0.6 | 701 ± 62 149 ± 137 3 ± 1.9 | 712 ± 59 151 ± 140 3.2 ± 2.2 | 741 ± 4 91 ± 50 3.5 ± 2.6 |
| Brain Regions | Test between MAB and HAB Groups Using t-Test | Test Corrected for Multiple Regions Comparison Using Bonferroni Adjustment | Test between HC, PD and lrrk2-UC in MAB Groups Using ANOVA Followed by Post Hoc Test | Test Corrected for Multiple Regions Comparison Using Bonferroni Adjustment | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HC | PD | UC | HC | PD | UC | HC/PD | HC/UC | HC/PD | HC/UC | |
| ROI | p values | |||||||||
| Acing | <0.001 | 0.03 | 0.02 | 0.00001 | 0.09 | 0.06 | 0.02 | 0.01 | 0.15 | 0.07 |
| Amygdala | <0.001 | 0.36 | 0.05 | 0.00019 | 0.39 | 0.07 | 0.13 | 0.1 | 0.18 | 0.11 |
| Anterior_Frontal | <0.001 | 0.01 | 0.02 | 0.00007 | 0.09 | 0.06 | 0.07 | 0.02 | 0.15 | 0.07 |
| Caudate | <0.001 | 0.04 | 0.05 | 0.00008 | 0.09 | 0.07 | 0.13 | 0.07 | 0.18 | 0.09 |
| DLPFC | <0.001 | 0.01 | 0.01 | 0.00002 | 0.09 | 0.05 | 0.04 | 0.03 | 0.15 | 0.07 |
| Dentate_Nucleus | <0.001 | 0.15 | 0.24 | 0.00001 | 0.20 | 0.25 | 0.12 | 0.03 | 0.18 | 0.07 |
| Globus_Pallidus | <0.001 | 0.21 | 0.09 | 0.00001 | 0.24 | 0.11 | 0.05 | 0.07 | 0.15 | 0.09 |
| Hypothalamus | <0.001 | 0.17 | 0.09 | 0.00014 | 0.21 | 0.11 | 0.09 | 0.03 | 0.15 | 0.07 |
| Insula | <0.001 | 0.03 | 0.04 | 0.00007 | 0.09 | 0.07 | 0.22 | 0.06 | 0.25 | 0.09 |
| Medulla | <0.001 | 0.17 | 0.96 | 0.00034 | 0.21 | 0.96 | 0.51 | 0.01 | 0.51 | 0.07 |
| Midbrain | <0.001 | 0.14 | 0.02 | 0.00046 | 0.20 | 0.06 | 0.34 | 0.32 | 0.37 | 0.32 |
| OFC | <0.001 | 0.09 | 0.04 | 0.00001 | 0.15 | 0.07 | 0.04 | 0.04 | 0.15 | 0.07 |
| Pcing | <0.001 | 0.04 | 0.02 | 0.00001 | 0.09 | 0.06 | 0.08 | 0.04 | 0.15 | 0.07 |
| PPN | <0.001 | 0.51 | 0.10 | 0.00004 | 0.51 | 0.12 | 0.51 | 0.21 | 0.51 | 0.21 |
| Parietal | <0.001 | 0.02 | 0.03 | 0.00001 | 0.09 | 0.06 | 0.02 | 0.006 | 0.15 | 0.07 |
| Pons | <0.001 | 0.12 | 0.10 | 0.00034 | 0.19 | 0.12 | 0.16 | 0.03 | 0.21 | 0.07 |
| Putamen | <0.001 | 0.04 | 0.004 | 0.00002 | 0.09 | 0.05 | 0.08 | 0.07 | 0.15 | 0.09 |
| SN | <0.001 | 0.38 | 0.12 | 0.00032 | 0.40 | 0.13 | 0.21 | 0.09 | 0.25 | 0.10 |
| Temporal | <0.001 | 0.05 | 0.01 | 0.00001 | 0.09 | 0.06 | 0.06 | 0.03 | 0.15 | 0.07 |
| Thalamus | <0.001 | 0.04 | 0.03 | 0.00001 | 0.09 | 0.06 | 0.09 | 0.07 | 0.15 | 0.09 |
| VS | <0.001 | 0.06 | 0.0006 | 0.00001 | 0.09 | 0.004 | 0.04 | 0.05 | 0.15 | 0.08 |
| Cerebellum | <0.001 | 0.02 | 0.05 | 0.00004 | 0.09 | 0.07 | 0.05 | 0.02 | 0.15 | 0.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mabrouk, R. Principal Component Analysis versus Subject’s Residual Profile Analysis for Neuroinflammation Investigation in Parkinson Patients: A PET Brain Imaging Study. J. Imaging 2022, 8, 56. https://doi.org/10.3390/jimaging8030056
Mabrouk R. Principal Component Analysis versus Subject’s Residual Profile Analysis for Neuroinflammation Investigation in Parkinson Patients: A PET Brain Imaging Study. Journal of Imaging. 2022; 8(3):56. https://doi.org/10.3390/jimaging8030056
Chicago/Turabian StyleMabrouk, Rostom. 2022. "Principal Component Analysis versus Subject’s Residual Profile Analysis for Neuroinflammation Investigation in Parkinson Patients: A PET Brain Imaging Study" Journal of Imaging 8, no. 3: 56. https://doi.org/10.3390/jimaging8030056