

pH-Sensitive Poly(acrylic acid)-g-poly(L-lysine) Charge-Driven Self-Assembling Hydrogels with 3D-Printability and Self-Healing Properties



Abstract
:
1. Introduction
2. Results and Discussion
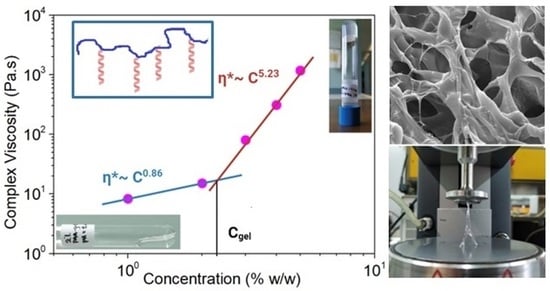
2.1. Self-Assembling Hydrogel
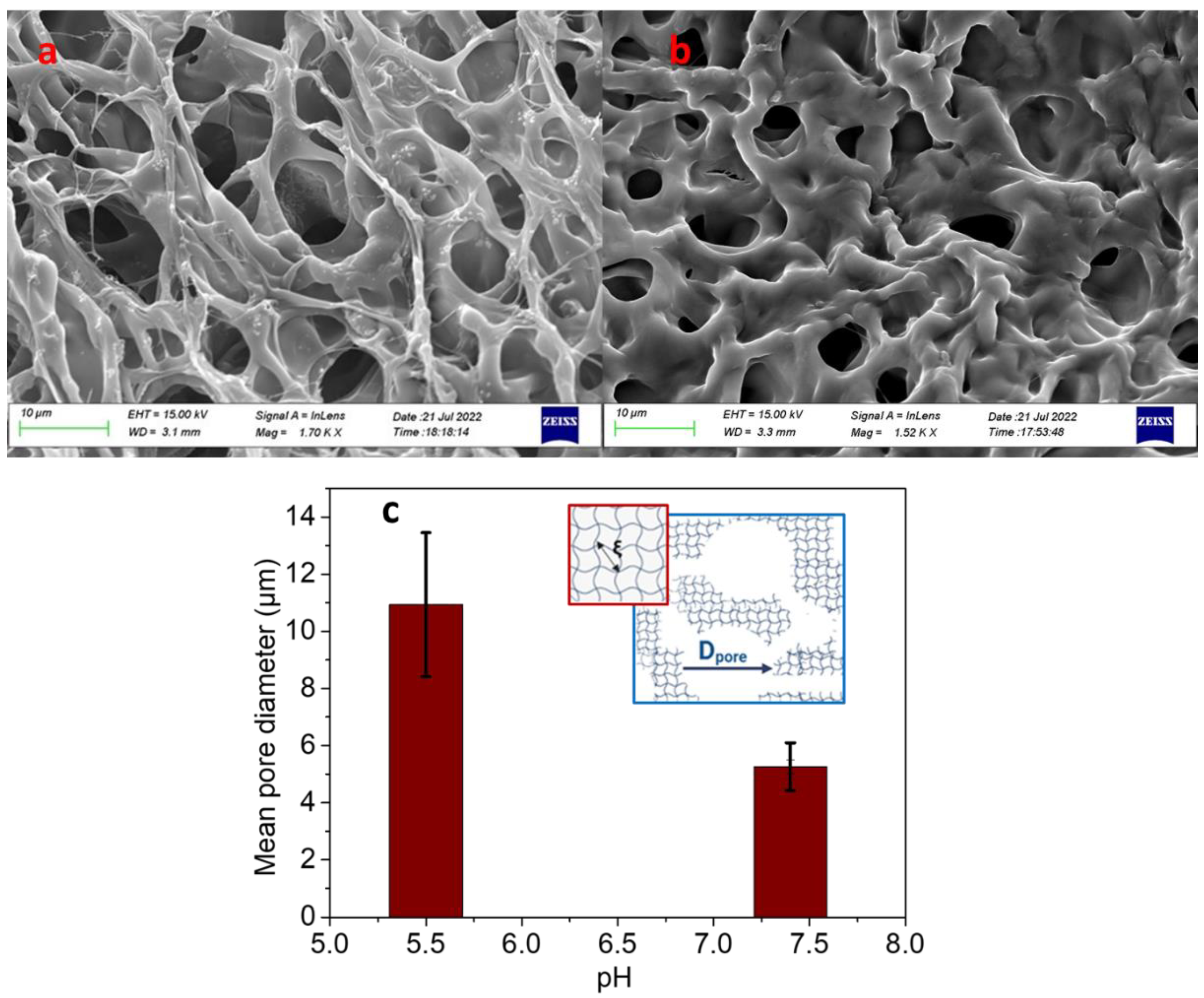
2.2. Effect of pH
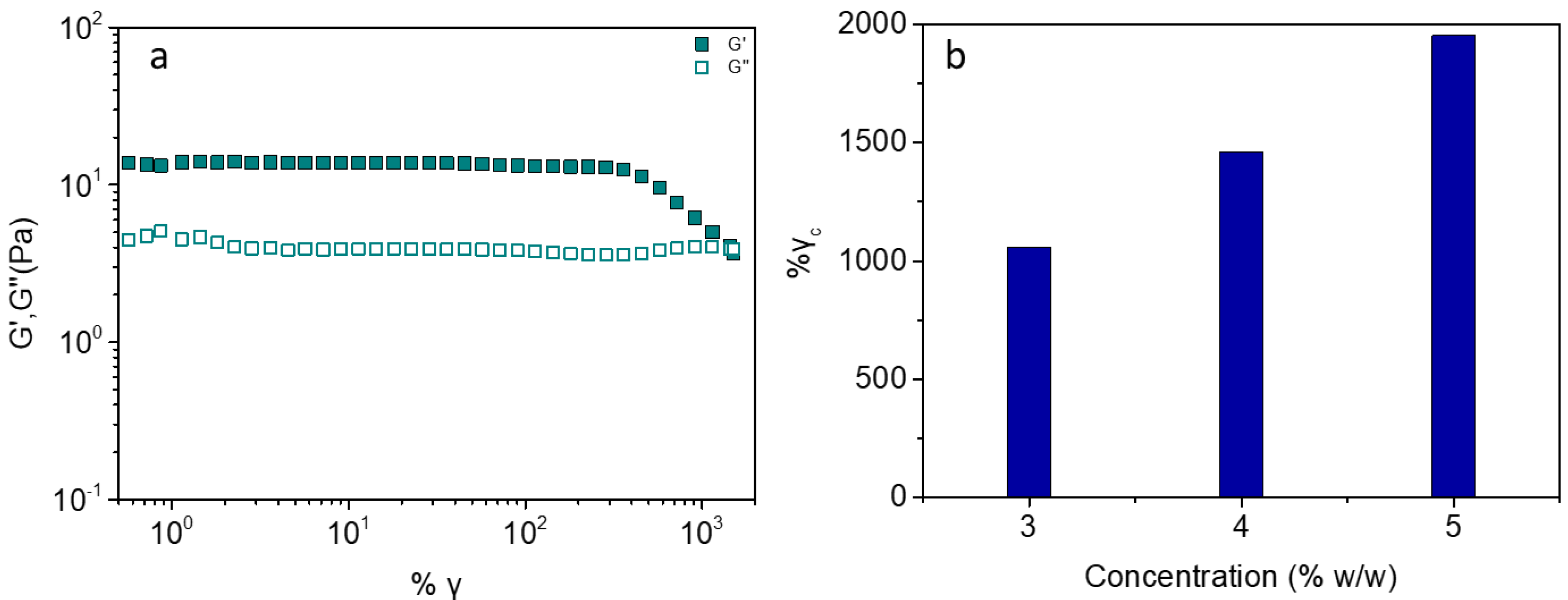
2.3. Effect of Ionic Strength
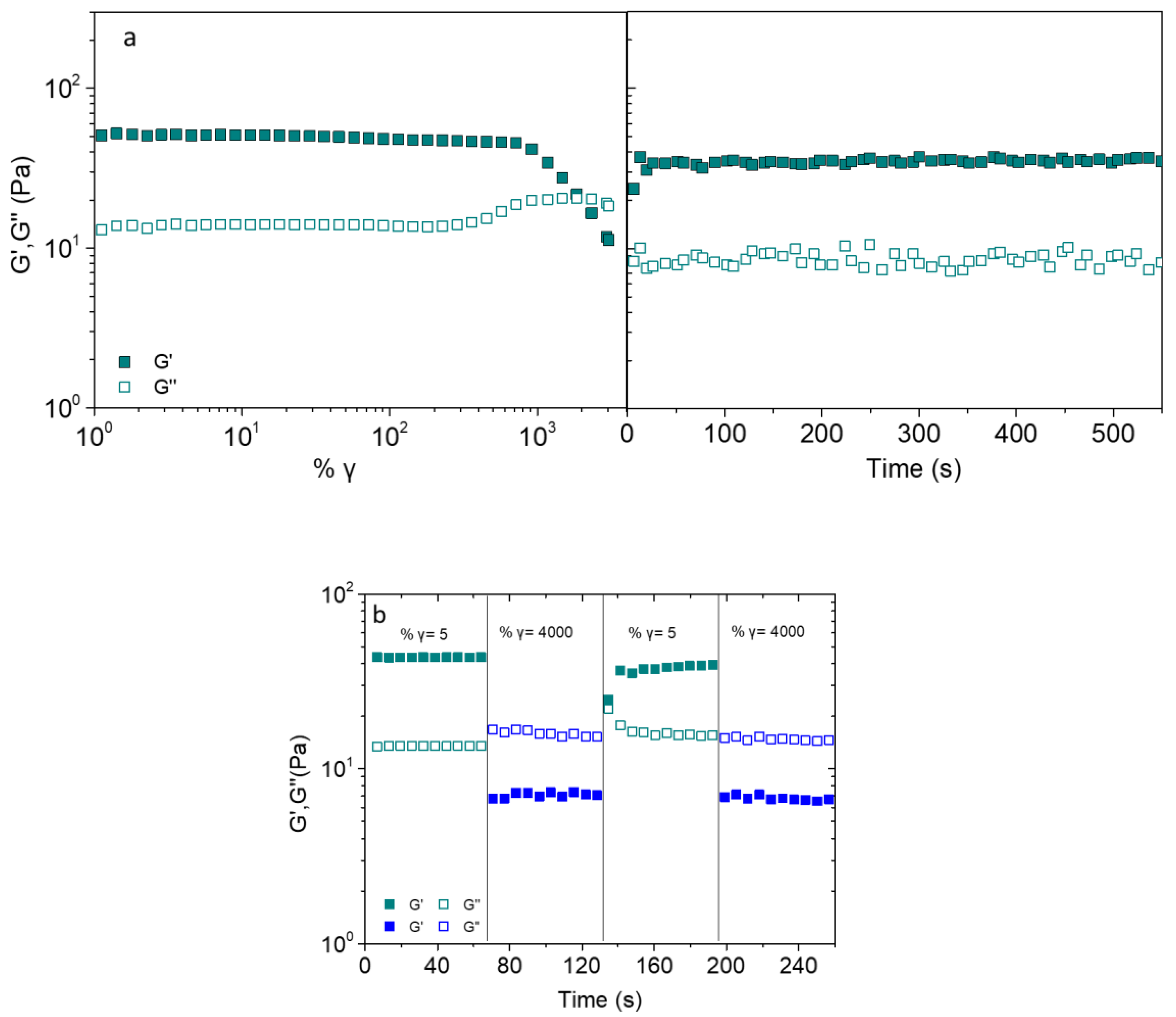
2.4. Shear-Induced Reversibility, Self-Healing and 3D-Printability
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Synthesis and Characterization
4.3. Preparation of samples for 1H-NMR Spectroscopy
4.4. Hydrogel Sample Preparation
4.5. Rheology
4.6. Scanning Electron Microscopy
4.7. Circular Dichroism
4.8. Zeta Potential
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koetting, M.C.; Peters, J.T.; Steichen, S.D.; Peppas, N.A. Stimulus-responsive hydrogels: Theory, modern advances, and applications. Mater. Sci. Eng. R 2015, 93, 1–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Feng, Q.; Fang, Z.; Gu, L.; Bian, L. Structurally dynamic hydrogels for biomedical applications: Pursuing a fine balance between macroscopic stability and microscopic dynamics. Chem. Rev. 2021, 121, 11149–11193. [Google Scholar] [CrossRef]
- Bashiri, S.; Hina, M.; Iqbal, J.; Rajpar, A.H.; Mujtaba, M.A.; Alghamdi, N.A.; Wageh, S.; Ramesh, K.; Ramesh, S. Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications. Polymers 2020, 12, 2702. [Google Scholar] [CrossRef] [PubMed]
- Constantinou, A.P.; Georgiou, T.K. Pre-clinical and clinical applications of thermoreversible hydrogels in biomedical engineering: A review. Polym. Int. 2021, 70, 1433–1448. [Google Scholar] [CrossRef]
- Li, Y.; Rodrigues, J.; Tomás, H. Injectable and biodegradable hydrogels: Gelation, biodegradation and biomedical applications. Chem. Soc. Rev. 2012, 41, 2193–2221. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, X.; Yuk, H.; Lin, S.; Liu, X.; Parada, G. Soft materials by design: Unconventional polymer networks give extreme properties. Chem. Rev. 2021, 121, 4309–4372. [Google Scholar] [CrossRef]
- Tsitsilianis, C. Responsive reversible hydrogels from associative “smart” macromolecules. Soft Matter 2010, 6, 2372–2388. [Google Scholar] [CrossRef]
- Papadakis, M.C.; Tsitsilianis, C. Responsive Hydrogels from Associative Block Copolymers: Physical Gelling through Polyion Complexation. Gels 2017, 3, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.L.; Kurokawa, T.; Kuroda, S.; Ihsan, A.B.; Akasaki, T.; Sato, K.; Haque, M.A.; Nakajima, T.; Gong, J.P. Physical Hydrogels Composed of Polyampholytes Demonstrate High Toughness and Viscoelasticity. Nat. Mater. 2013, 12, 932–937. [Google Scholar] [CrossRef] [Green Version]
- Bossard, F.; Sfika, V.; Tsitsilianis, C. Rheological properties of physical gel formed by triblock polyampholyte in salt-fee aqueous solutions. Macromolecules 2004, 37, 3899–3904. [Google Scholar] [CrossRef]
- Dyakonova, M.A.; Stavrouli, N.; Popescu, M.T.; Kyriakos, K.; Grillo, I.; Philipp, M.; Jaksch, S.; Tsitsilianis, C.; Papadakis, C.M. Physical hydrogels via charge driven self-organization of a triblock polyampholyte–rheological and structural investigations. Macromolecules 2014, 47, 7561–7572. [Google Scholar] [CrossRef]
- Dyakonova, M.A.; Berezkin, A.V.; Kyriakos, K.; Gkermpoura, S.; Popescu, M.T.; Filippov, S.K.; Štěpánek, P.; Di, Z.; Tsitsilianis, C.; Papadakis, C.M. Salt-induced changes in triblock polyampholyte hydrogels—Computer simulations, rheological, structural and dynamic characterization. Macromolecules 2015, 48, 8177–8189. [Google Scholar] [CrossRef]
- Bossard, F.; Tsitsilianis, C.; Yannopoulos, S.N.; Petekidis, G.; Sfika, V. A Novel Thermothickening Phenomenon Exhibited by a Triblock Polyampholyte in Aqueous Salt-Free Solutions. Macromolecules 2005, 38, 2883–2888. [Google Scholar] [CrossRef]
- Tsianou, M.; KjØniksen, A.-L.; Thuresson, K.; NystrÖm, B. Light scattering and viscoelasticity in aqueous mixtures of oppositely charged and hydrophobically modified polyelectrolutes. Macromolecules 1999, 32, 2974–2982. [Google Scholar] [CrossRef]
- Lemmers, M.; Sprakel, J.; Voets, I.K.; van der Gucht, J.; Cohen Stuart, M.A. Multiresponsive reversible gels based on charge-driven assembly. Angew. Chem. Int. Ed. 2010, 49, 708–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, S.; Kaneko, J.; Nagasaki, Y. Dual stimuli-responsive redox-active injectable gel by polyion complex based flower micelles for biomedical applications. Macromolecules 2015, 48, 3088–3094. [Google Scholar] [CrossRef]
- Hunt, J.N.; Feldman, K.E.; Lynd, N.A.; Deek, J.; Campos, L.M.; Spruell, J.M.; Hernandez, B.M.; Kramer, E.J.; Hawker, C.J. Tunable, high-modulus hydrogels driven by ionic coacervation. Adv. Mater. 2011, 23, 2327–2331. [Google Scholar] [CrossRef]
- Srivastava, S.; Levi, A.E.; Goldfeld, D.J.; and Tirrell, M.V. Structure, Morphology, and Rheology of Polyelectrolyte Complex Hydrogels Formed by Self-Assembly of Oppositely Charged Triblock Polyelectrolytes. Macromolecules 2020, 53, 5763–5774. [Google Scholar] [CrossRef]
- Kim, S.; Kim, J.-M.; Wood, K.; Choi, S.-H. Ionic group-dependent structure of complex coacervate hydrogels formed by ABA triblock copolymers. Soft Matter 2022, 18, 4146–4155. [Google Scholar] [CrossRef]
- Cross, D.; Jiang, X.; Ji, W.; Han, W.; Wang, C. Injectable hydrogels of hyaluronic acid crosslinked by wll-defined synthetic polycations: Preparation and characterization in vitro and in vivo. Macromol. Biosci. 2015, 15, 668–681. [Google Scholar] [CrossRef]
- Kim, J.-M.; Heo, T.-Y.; Choi, S.-H. Structure and Relaxation Dynamics for Complex Coacervate Hydrogels Formed by ABA Triblock Copolymers. Macromolecules 2020, 53, 9234–9243. [Google Scholar] [CrossRef]
- Stano, R.; Kosovan, P.; Tagliabue, A.; Holm, C. Electrostatically cross-linked reversible gels—Effects of pH and ionic strength. Macromolecules 2012, 54, 4769–4781. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravanou, S.F.; Ioannidis, K.; Dimopoulos, A.; Paxinou, A.; Kounelaki, F.; Varsami, S.M.; Tsitsilianis, C.; Papantoniou, I.; Pasparakis, G. Dually Crosslinked Injectable Alginate-Based Graft Copolymer Thermo-Responsive Hydrogels as 3D Printing Bioinks for Cell Spheroid Growth and Release. Carbohydr. Polym. 2023, 312, 120790. [Google Scholar] [CrossRef] [PubMed]
- Chassenieux, C.; Tsitsilianis, C. Recent trends on pH/thermo-responsive self-assembling hydrogels: From polyions to peptide-based polymeric gelators. Soft Matter 2016, 12, 1344–1359. [Google Scholar] [CrossRef] [PubMed]
- Rosa, E.; Gallo, E.; Sibillano, T.; Giannini, C.; Rizzuti, S.; Gianolio, E.; Scognamiglio, P.L.; Morelli, G.; Accardo, A.; Diaferia, C. Incorporation of PEG Diacrylates (PEGDA) Generates Hybrid Fmoc-FF Hydrogel Matrices. Gels 2022, 8, 831. [Google Scholar] [CrossRef]
- Diaferia, C.; Rosa, E.; Balasco, N.; Sibillano, T.; Morelli, G.; Giannini, C.; Vitagliano, L.; Accardo, A. The introduction of a cysteine residue modulates the mechanical properties of aromatic-based solid aggregates and self-supporting hydrogels. Chem. Eur. J. 2021, 27, 14886–14898. [Google Scholar] [CrossRef]
- Dharmayanti, C.; Gillam, T.A.; Klingler-Hoffmann, M.; Albrecht, H.; Blencowe, A. Strategies for the Development of pH-Responsive Synthetic Polypeptides and Polymer-Peptide Hybrids: Recent Advancements. Polymers 2021, 13, 624. [Google Scholar] [CrossRef]
- Ji, D.-Y.; Kuob, T.-F.; Wua, H.-D.; Yangc, J.-C.; Lee, S.-Y. A novel injectable chitosan/polyglutamate polyelectrolyte complex hydrogel with hydroxyapatite for soft-tissue augmentation. Carbohydr. Polym. 2012, 89, 1123–1130. [Google Scholar] [CrossRef]
- Bonduelle, C. Secondary structures of synthetic polypeptide polymers. Polym. Chem. 2018, 9, 1517–1529. [Google Scholar] [CrossRef]
- Popescu, M.-T.; Liontos, G.; Avgeropoulos, Q.A.; Tsitsilianis, C. Stimuli Responsive Fibrous Hydrogels from Hierarchical Self-Assembly of a Triblock Copolypeptide. Soft Matter 2015, 11, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Oatari, Y.; Motoda, H.; Nishimura, S.; Sasaki, Y.; Okamoto, Y.; Yamamoto, D.; Shioi, A.; Higashi, N. Star-Shaped Peptide−Polymer Hybrids as Fast pH-Responsive Supramolecular Hydrogels. Biomacromolecules 2022, 23, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wollenberg, A.L.; O’Shea, T.M.; Cui, Y.; Zhou, Z.H.; Sofroniew, M.V.; Deming, T.J. Conformation-Directed Formation of Self-Healing Diblock Copolypeptide Hydrogels via Polyion Complexation. J. Am. Chem. Soc. 2017, 139, 15114–15121. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Zhuang, X.; He, C.; Wei, Y.; Chen, X. High performance and reversible ionic polypeptide hydrogel based on charge-driven assembly for biomedical applications. Acta Biomater. 2015, 11, 183–190. [Google Scholar] [CrossRef]
- Karga, M.-E.; Kargaki, M.-E.; Iatrou, H.; Tsitsilianis, C. pH-Responsive, Thermo-Resistant Poly(Acrylic Acid)-g-Poly(boc-L-Lysine) Hydrogel with Shear-Induced Injectability. Gels 2022, 8, 817. [Google Scholar] [CrossRef]
- Krause, W.E.; Bellomo, E.G.; Colby, R.H. Rheology of sodium hyaluronate under physiological conditions. Biomacromolecules 2001, 2, 65–69. [Google Scholar] [CrossRef]
- Stavrouli, N.; Aubry, T.C. Rheological Properties of ABA Telechelic Polyelectrolyte and ABA Polyampholyte Reversible Hydrogels: A Comparative Study. Polymer 2008, 49, 1249–1256. [Google Scholar] [CrossRef]
- Colby, R.H. Structure and linear viscoelasticity of flexible polymer solutions: Comparison of polyelectrolyte and neutral polymer solutions. Rheol. Acta 2010, 49, 425–442. [Google Scholar] [CrossRef]
- Krogstad, D.V.; Lynd, N.A.; Choi, S.-H.; Spruell, J.M.; Hawker, C.J.; Kramer, E.J.; Tirrell, M.V. Effects of polymer and salt concentration on the structure and properties of triblock coacervate hydrogels. Macromolecules 2013, 46, 1512–1518. [Google Scholar] [CrossRef]
- Landsgesell, J.; Nova, L.; Rud, O.; Uhlik, F.; Sean, D.; Hebbeker, P.; Holm, C.; Košovan, P. Simulations of Ionization Equilibria in Weak Polyelectrolyte Solutions and Gels. Soft Matter 2019, 15, 1155–1185. [Google Scholar] [CrossRef]
- Myer, Y.P. The pH-Induced Helix-Coil Transition of Poly-L-Lysine and Poly-L-Glutamic Acid and the 238-mμ Dichroi Band. Macromolecules 1969, 2, 624–628. [Google Scholar] [CrossRef]
- Grigsby, J.J.; Blanch, H.W.; Prausnitz, J.M. Effect of secondary structure on the potential of mean force for poly-L-lysine in the α-helix and β-sheet conformations. Biophys. Chem. 2002, 99, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.C.; Bonner, D.K.; Buss, H.G.; Cheunga, E.Y.; Hammond, P.T. The synthetic tuning of clickable pH responsive cationic polypeptides and block copolypeptides. Soft Matter 2011, 7, 5627–5637. [Google Scholar] [CrossRef]
- Shinoda, K.; Hayashi, T.; Yoshida, T.; Sakai, K.; Nakajima, A. Complex formation of Poly-L-lysine with Poly(acrylic acid). Polymer J. 1976, 8, 202–207. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Boswell, P.G.; Bühlmann, P.; Lodge, T.P. Ion Gels by Self-Assembly of a Triblock Copolymer in an Ionic Liquid. J. Phys. Chem. B 2007, 111, 4645–4652. [Google Scholar] [CrossRef]
- Tsitsilianis, C.; Serras, G.; Ko, C.-H.; Jung, F.; Papadakis, C.-M.; Rikkou-Kalourkoti, M.; Patrickios, C.S.; Schweins, R.; Chassenieux, C. Thermoresponsive Hydrogels Based on Telechelic Polyelectrolytes: From Dynamic to “Frozen” Networks. Macromolecules 2018, 51, 2169–2179. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | CP (wt%) | pH | Ionic Strength (M) |
|---|---|---|---|
| 1 | 1 | 7.4 | - |
| 2 | 2 | 7.4 | - |
| 3 | 3 | 7.4 | - |
| 4 | 4 | 7.4 | - |
| 5 | 5 | 7.4 | - |
| 6 | 5 | 5.5 | - |
| 7 | 5 | 9.0 | - |
| 8 | 5 | 11.9 | - |
| 9 | 5 | 7.4 | 0.15 |
| 10 | 5 | 7.4 | 0.30 |
| 11 | 5 | 7.4 | 0.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kargaki, M.-E.; Arfara, F.; Iatrou, H.; Tsitsilianis, C. pH-Sensitive Poly(acrylic acid)-g-poly(L-lysine) Charge-Driven Self-Assembling Hydrogels with 3D-Printability and Self-Healing Properties. Gels 2023, 9, 512. https://doi.org/10.3390/gels9070512
Kargaki M-E, Arfara F, Iatrou H, Tsitsilianis C. pH-Sensitive Poly(acrylic acid)-g-poly(L-lysine) Charge-Driven Self-Assembling Hydrogels with 3D-Printability and Self-Healing Properties. Gels. 2023; 9(7):512. https://doi.org/10.3390/gels9070512
Chicago/Turabian StyleKargaki, Maria-Eleni, Foteini Arfara, Hermis Iatrou, and Constantinos Tsitsilianis. 2023. "pH-Sensitive Poly(acrylic acid)-g-poly(L-lysine) Charge-Driven Self-Assembling Hydrogels with 3D-Printability and Self-Healing Properties" Gels 9, no. 7: 512. https://doi.org/10.3390/gels9070512