
From Macro to Micro: Comparison of Imaging Techniques to Detect Vascular Network Formation in Left Ventricle Decellularized Extracellular Matrix Hydrogels
 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Vascular Structure Is Shown by Inverted Fluorescent Microscopy
2.1.2. Vascular Structure in 3D Shown by the Live Cell Confocal Imaging System
2.1.3. Optical Coherence Tomography
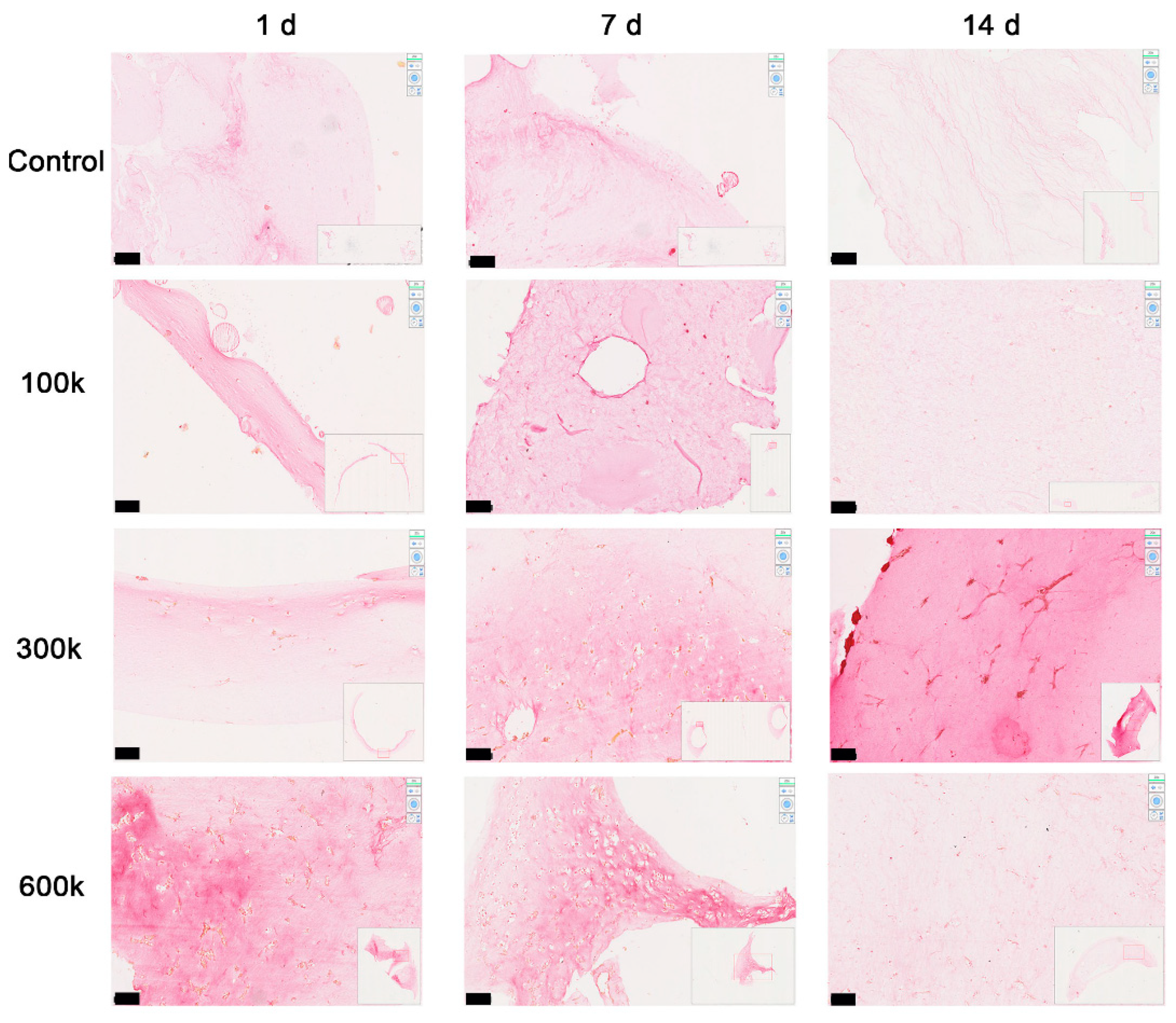
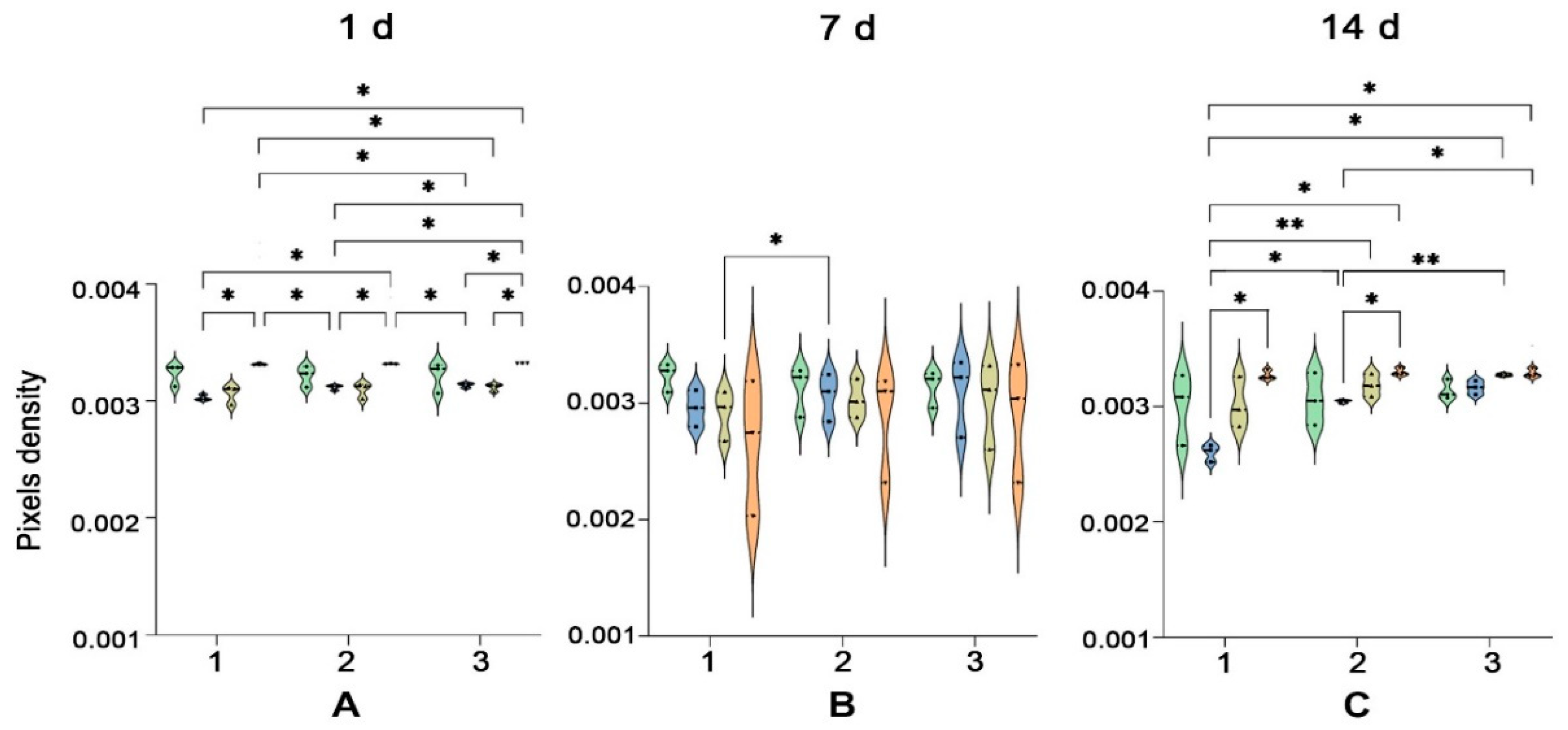
2.1.4. Vascular-like Structure Analyses by H&E Staining
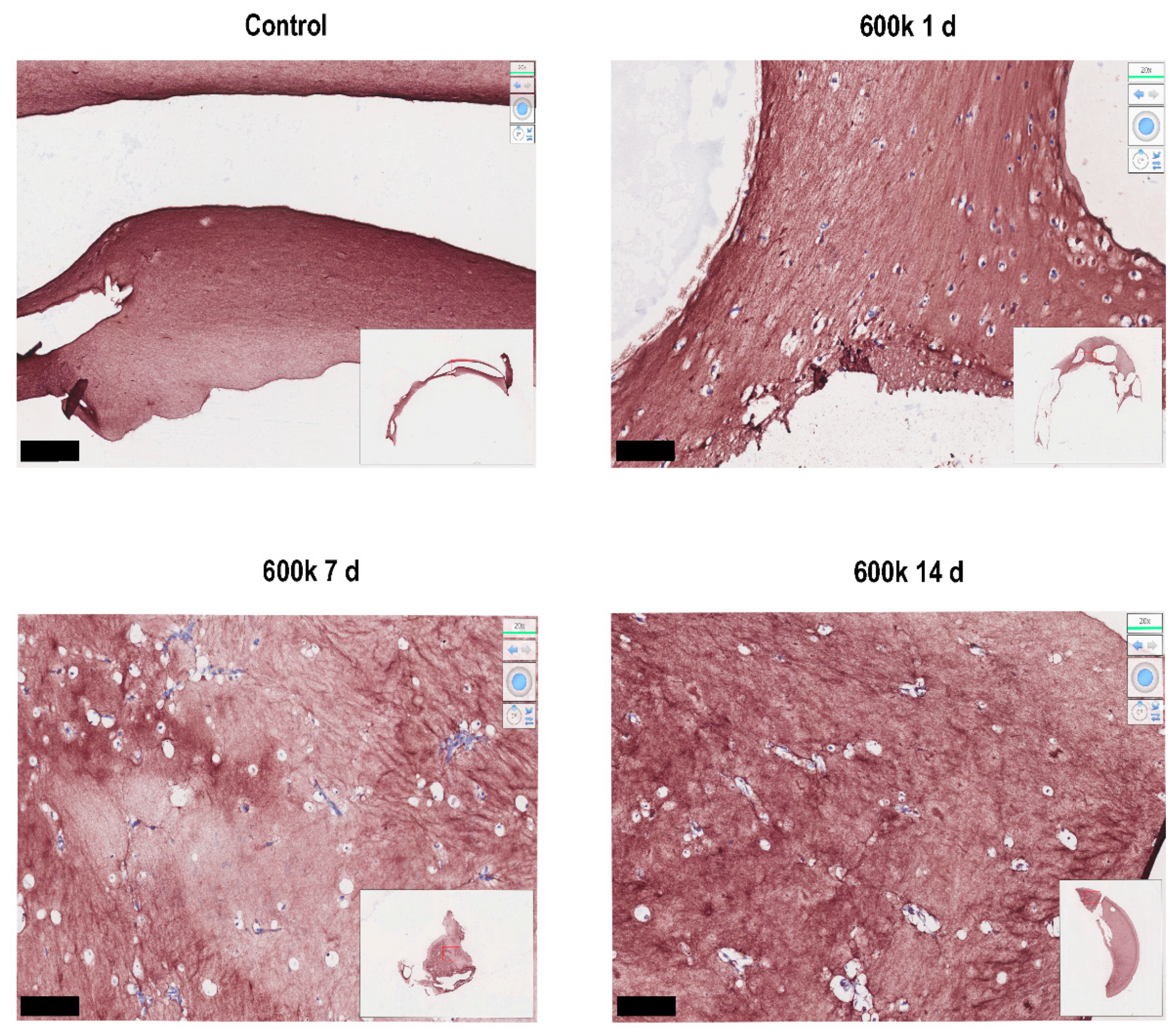
2.1.5. Collagen Is Deposited in the Close Vicinity of Vascular Networks
2.2. Discussion
2.2.1. Technical Clues
2.2.2. Cell Behavior in 3D Clues
3. Conclusions
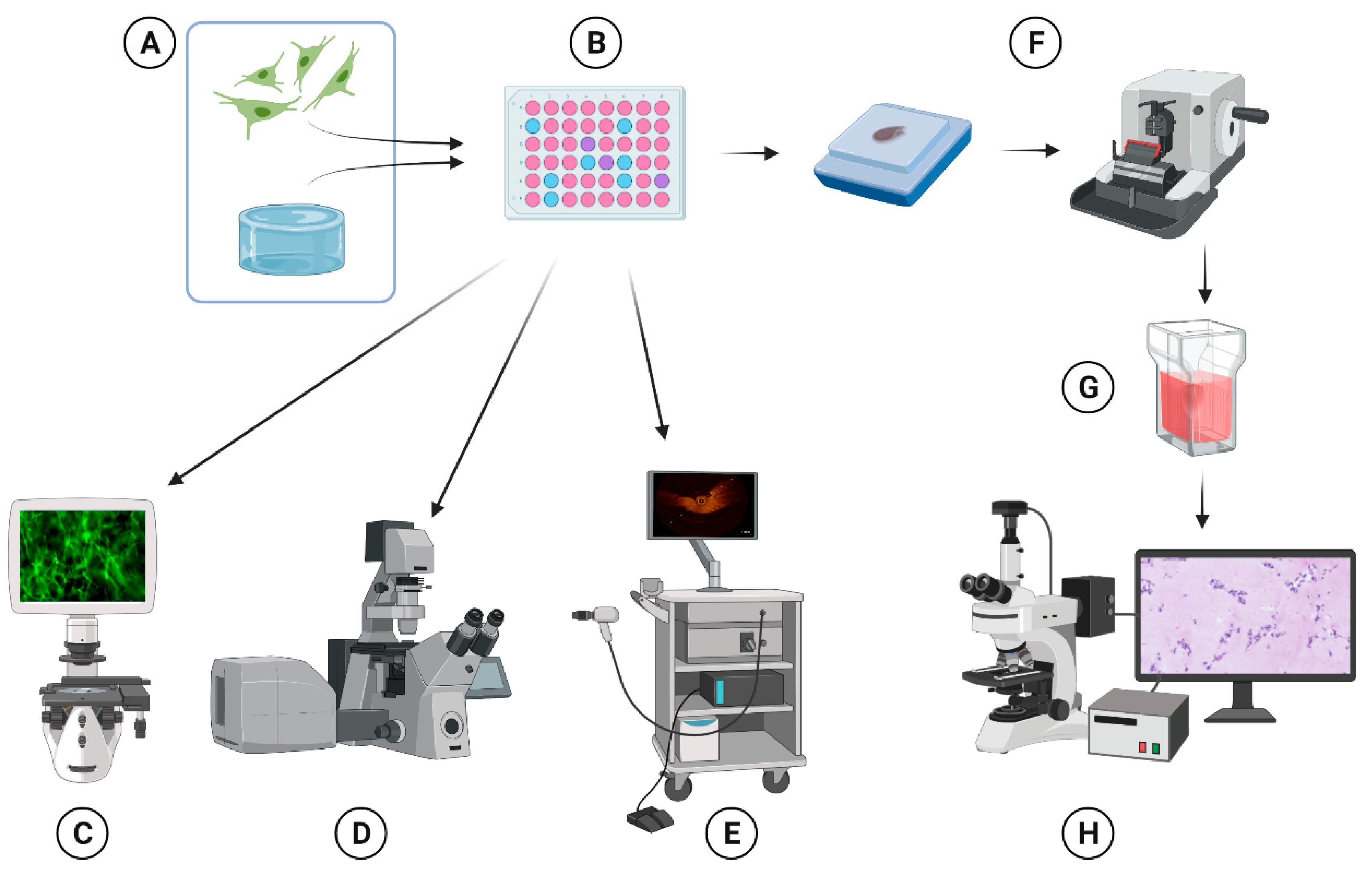
4. Materials and Methods
4.1. Hydrogel Synthesis
4.2. 3D Cell Culture
4.3. Fluorescent Microscopy (2D)
4.4. Live Cell Confocal Imaging System
4.5. Optical Coherence Tomography
4.6. Immunohistochemical Analyses
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kobayashi, J.; Kikuchi, A.; Aoyagi, T.; Okano, T. Cell sheet tissue engineering: Cell sheet preparation, harvesting/manipulation, and transplantation. J. Biomed. Mater. Res. Part A 2019, 107, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Monchaux, E.; Vermette, P. Effects of surface properties and bioactivation of biomaterials on endothelial cells. Front. Biosci. 2010, 2, 239–255. [Google Scholar] [CrossRef] [Green Version]
- Nikolova, M.P.; Chavali, M.S. Recent advances in biomaterials for 3D scaffolds: A review. Bioact. Mater. 2019, 4, 271–292. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome—An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dongen, J.A.; Getova, V.; Brouwer, L.A.; Liguori, G.R.; Sharma, P.K.; Stevens, H.P.; van der Lei, B.; Harmsen, M.C. Adipose tissue-derived extracellular matrix hydrogels as a release platform for secreted paracrine factors. J. Tissue Eng. Regen. Med. 2019, 13, 973–985. [Google Scholar] [CrossRef] [Green Version]
- Liguori, G.R.; Liguori, T.T.A.; de Moraes, S.R.; Sinkunas, V.; Terlizzi, V.; van Dongen, J.A.; Sharma, P.K.; Moreira, L.F.P.; Harmsen, M.C. Molecular and Biomechanical Clues From Cardiac Tissue Decellularized Extracellular Matrix Drive Stromal Cell Plasticity. Front. Bioeng. Biotechnol. 2020, 8, 520. [Google Scholar] [CrossRef]
- Terlizzi, V.; Kolibabka, M.; Burgess, J.K.; Hammes, H.P.; Harmsen, M.C. The Pericytic Phenotype of Adipose Tissue-Derived Stromal Cells Is Promoted by NOTCH2. Stem Cells 2018, 36, 240–251. [Google Scholar] [CrossRef] [Green Version]
- Fong, A.H.; Romero-López, M.; Heylman, C.M.; Keating, M.; Tran, D.; Sobrino, A.; Tran, A.Q.; Pham, H.H.; Fimbres, C.; Gershon, P.D.; et al. Three-Dimensional Adult Cardiac Extracellular Matrix Promotes Maturation of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Tissue Eng. Part A 2016, 22, 1016–1025. [Google Scholar] [CrossRef] [Green Version]
- Talaei-Khozani, T.; Yaghoubi, A. An overview of post transplantation events of decellularized scaffolds. Transpl. Immunol. 2022, 74, 101640. [Google Scholar] [CrossRef]
- Efraim, Y.; Sarig, H.; Cohen Anavy, N.; Sarig, U.; de Berardinis, E.; Chaw, S.Y.; Krishnamoorthi, M.; Kalifa, J.; Bogireddi, H.; Duc, T.V.; et al. Biohybrid cardiac ECM-based hydrogels improve long term cardiac function post myocardial infarction. Acta Biomater. 2017, 50, 220–233. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W.; Molema, G. Angiogenesis: Potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases, and chronic inflammation. Pharmacol. Rev. 2000, 52, 237–268. [Google Scholar] [PubMed]
- Marchand, M.; Monnot, C.; Muller, L.; Germain, S. Extracellular matrix scaffolding in angiogenesis and capillary homeostasis. Semin. Cell Dev. Biol. 2019, 89, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Raica, M.; Cimpean, A.M. Platelet-Derived Growth Factor (PDGF)/PDGF Receptors (PDGFR) Axis as Target for Antitumor and Antiangiogenic Therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Gordon, E.; Schimmel, L.; Frye, M. The Importance of Mechanical Forces for in vitro Endothelial Cell Biology. Front. Physiol. 2020, 11, 684. [Google Scholar] [CrossRef]
- Kim, J.W.; Nam, S.A.; Yi, J.; Kim, J.Y.; Lee, J.Y.; Park, S.Y.; Sen, T.; Choi, Y.M.; Lee, J.Y.; Kim, H.L.; et al. Kidney Decellularized Extracellular Matrix Enhanced the Vascularization and Maturation of Human Kidney Organoids. Adv. Sci. 2022, 9, e2103526. [Google Scholar] [CrossRef]
- Ettinger, A.; Wittmann, T. Fluorescence live cell imaging. Methods Cell Biol. 2014, 123, 77–94. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, Z.; Li, Y.; Yang, J.; Li, Y. Optical coherence tomography angiography for noninvasive evaluation of angiogenesis in a limb ischemia mouse model. Sci. Rep. 2019, 9, 5980. [Google Scholar] [CrossRef]
- Martinez-Garcia, F.D.; Valk, M.M.; Sharma, P.K.; Burgess, J.K.; Harmsen, M.C. Adipose Tissue-Derived Stromal Cells Alter the Mechanical Stability and Viscoelastic Properties of Gelatine Methacryloyl Hydrogels. Int. J. Mol. Sci. 2021, 22, 10153. [Google Scholar] [CrossRef]
- Martinez-Garcia, F.D.; van Dongen, J.A.; Burgess, J.K.; Harmsen, M.C. Matrix Metalloproteases from Adipose Tissue-Derived Stromal Cells Are Spatiotemporally Regulated by Hydrogel Mechanics in a 3D Microenvironment. Bioengineering 2022, 9, 340. [Google Scholar] [CrossRef]
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis Analyzer for ImageJ—A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2020, 10, 11568. [Google Scholar] [CrossRef]
- Carpaij, O.A.W.M.; Goorsenberg, D.M.; De Bruin, R.M.; Van Den Elzen, M.C.; Nawijn, H.A.M.; Kerstjens, M.; Van Den Berge, J.T.; Annema, J.K.; Burgess, P.I.B. Optical coherence tomography (OCT) detects collagen within the airway wall extracellular matrix. Eur. Respir. J. 2019, 54, PA3169. [Google Scholar]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Longair, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]










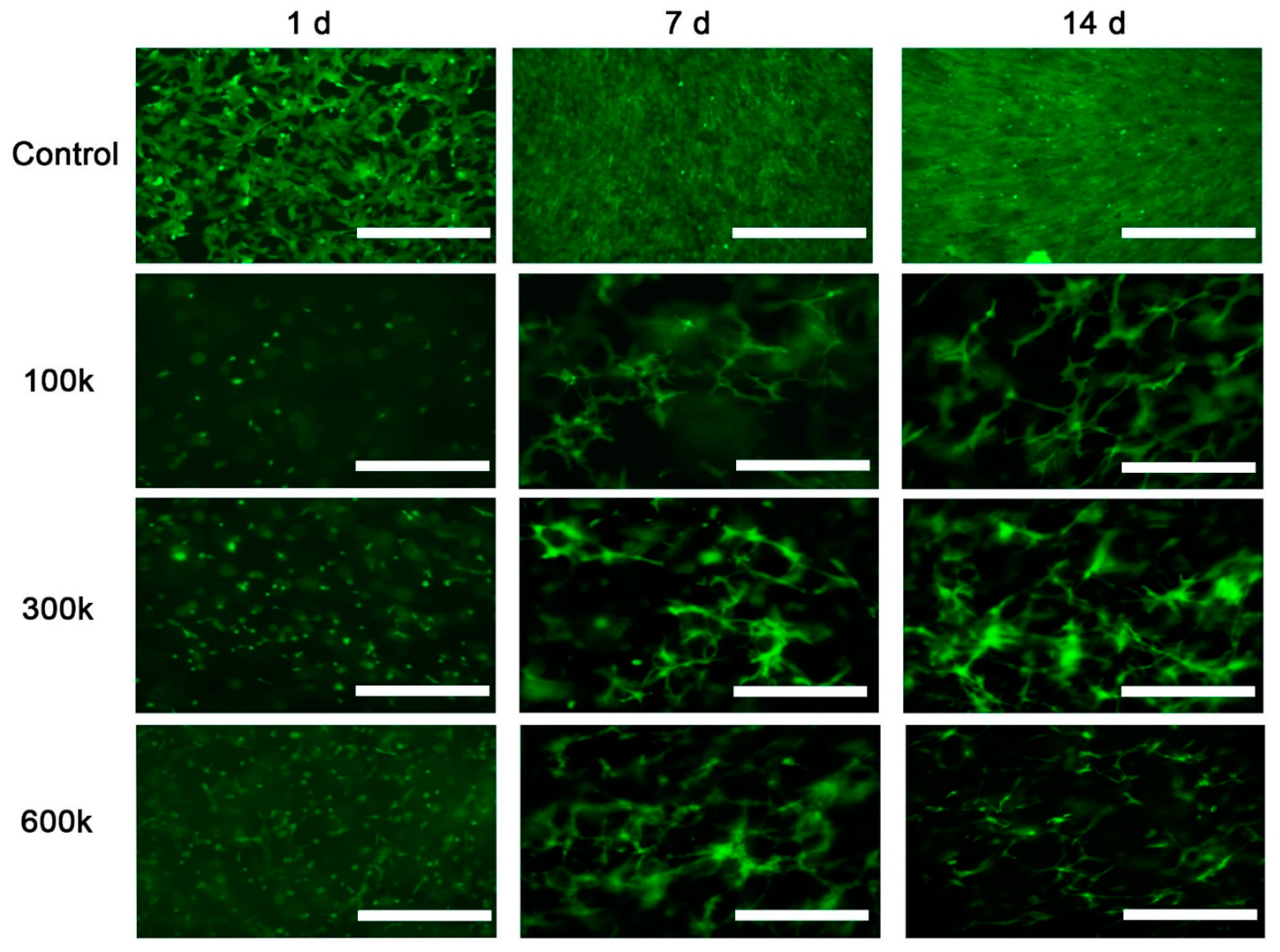{kind=link}
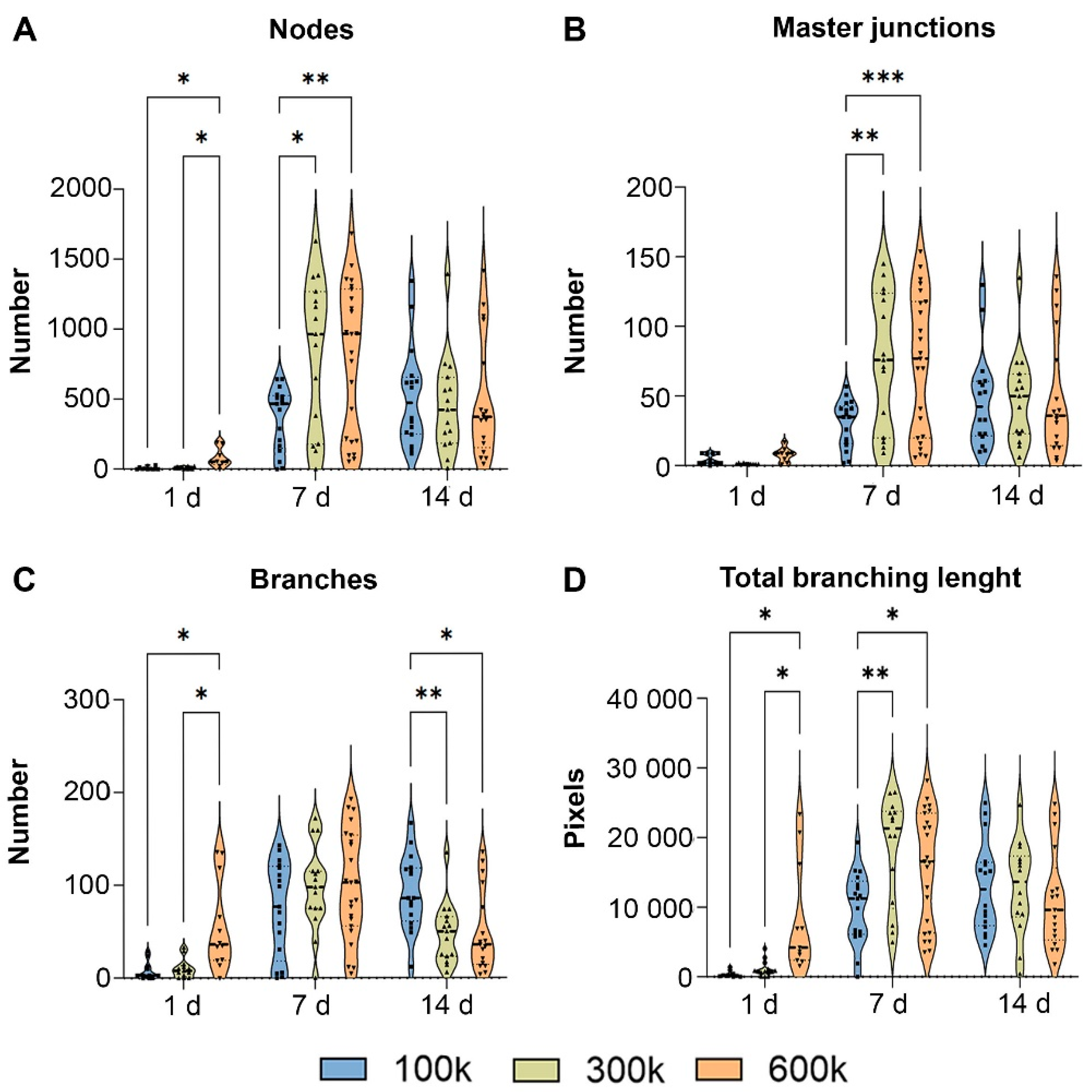{kind=link}
{kind=link}
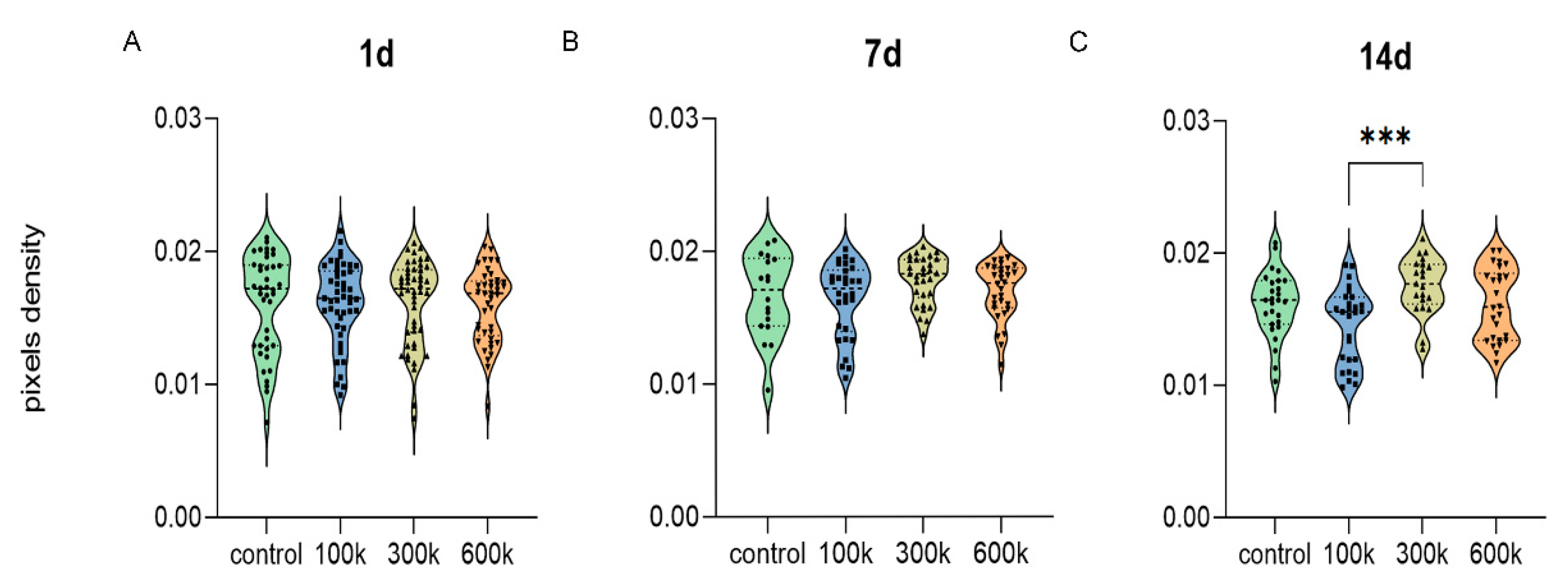{kind=link}
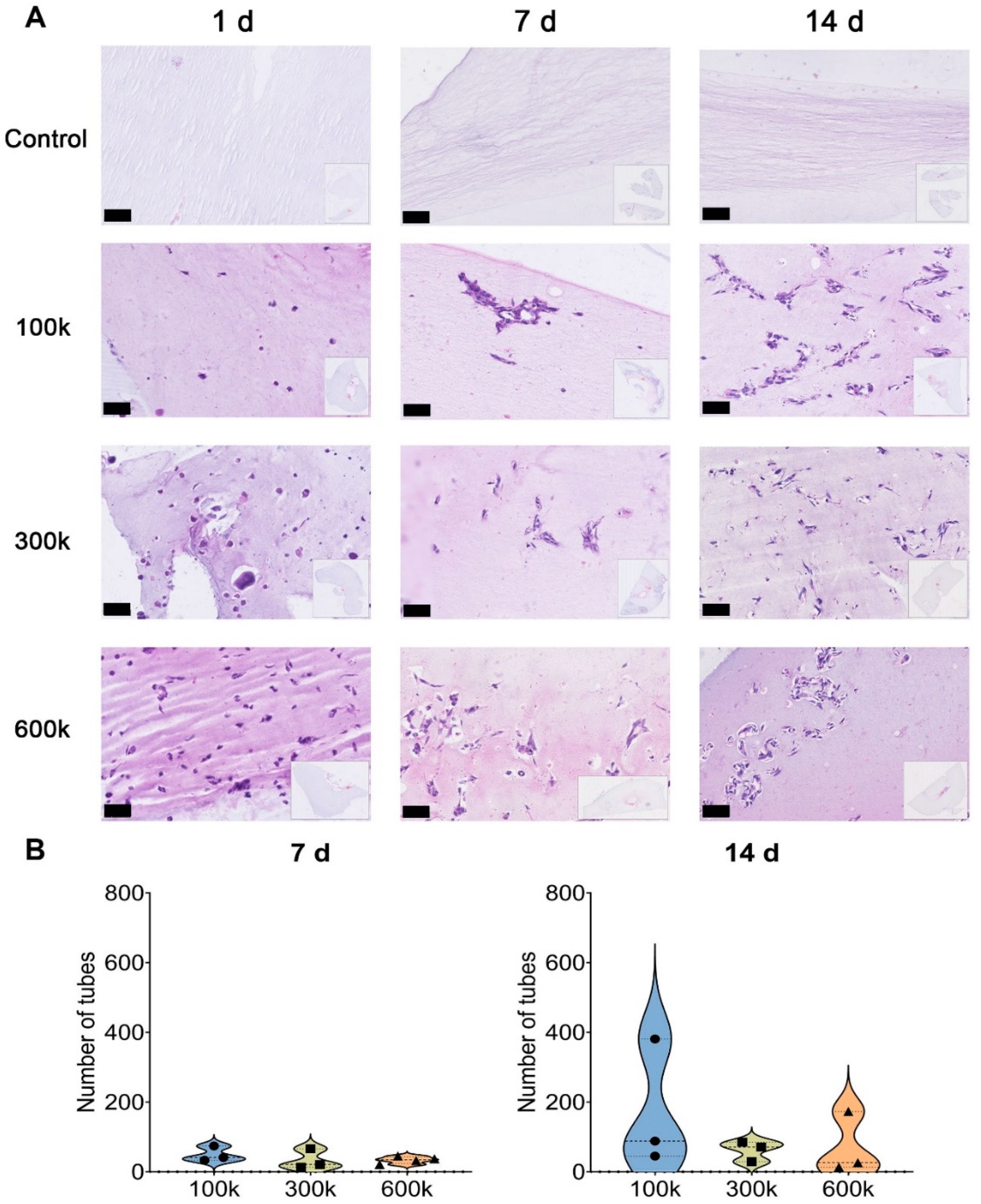{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Benefit | Limitation |
|---|---|---|
| Inverted fluorescence microscopy |
|
|
| Confocal microscopy |
|
|
| OCT |
|
|
| (Immuno)histochemistry |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Getova, V.E.; Martinez-Garcia, F.D.; Borghuis, T.; Burgess, J.K.; Harmsen, M.C. From Macro to Micro: Comparison of Imaging Techniques to Detect Vascular Network Formation in Left Ventricle Decellularized Extracellular Matrix Hydrogels. Gels 2022, 8, 729. https://doi.org/10.3390/gels8110729
Zhang M, Getova VE, Martinez-Garcia FD, Borghuis T, Burgess JK, Harmsen MC. From Macro to Micro: Comparison of Imaging Techniques to Detect Vascular Network Formation in Left Ventricle Decellularized Extracellular Matrix Hydrogels. Gels. 2022; 8(11):729. https://doi.org/10.3390/gels8110729
Chicago/Turabian StyleZhang, Meng, Vasilena E. Getova, Francisco Drusso Martinez-Garcia, Theo Borghuis, Janette K. Burgess, and Martin C. Harmsen. 2022. "From Macro to Micro: Comparison of Imaging Techniques to Detect Vascular Network Formation in Left Ventricle Decellularized Extracellular Matrix Hydrogels" Gels 8, no. 11: 729. https://doi.org/10.3390/gels8110729