
Unveiling Cryptic Species Diversity and Genetic Variation of Lasiodiplodia (Botryosphaeriaceae, Botryosphaeriales) Infecting Fruit Crops in Taiwan
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Single Spore Colony Isolation
2.2. DNA Extraction and Sequencing of Four Markers
2.3. Phylogenetic Analysis and Cryptic Species Analyses
2.4. Assessment of Nucleotide Diversity, Recombination, Linkage Disequilibrium and Demographic History
2.5. Genetic Differentiation and Genetic Structure Analysis
3. Results
3.1. Species Identification from Single Spore Colonies
3.2. Isolation Frequency (IF) Revealed That L. theobromae Has the Highest IF among Lasiodiplodia Species
3.3. Single Molecular Marker Does Not Provide Enough Resolution to Identify Species
3.4. Phylogenetic Analysis Using Combined Molecular Markers Confirmed the Deep Species Identification
3.5. Species Diversity Revealed in Lasiodiplodia Species
3.6. Genetic Diversity and Recombination Events in Lasiodiplodia Species
3.7. Significant Population Expansion Detected in L. theobromae
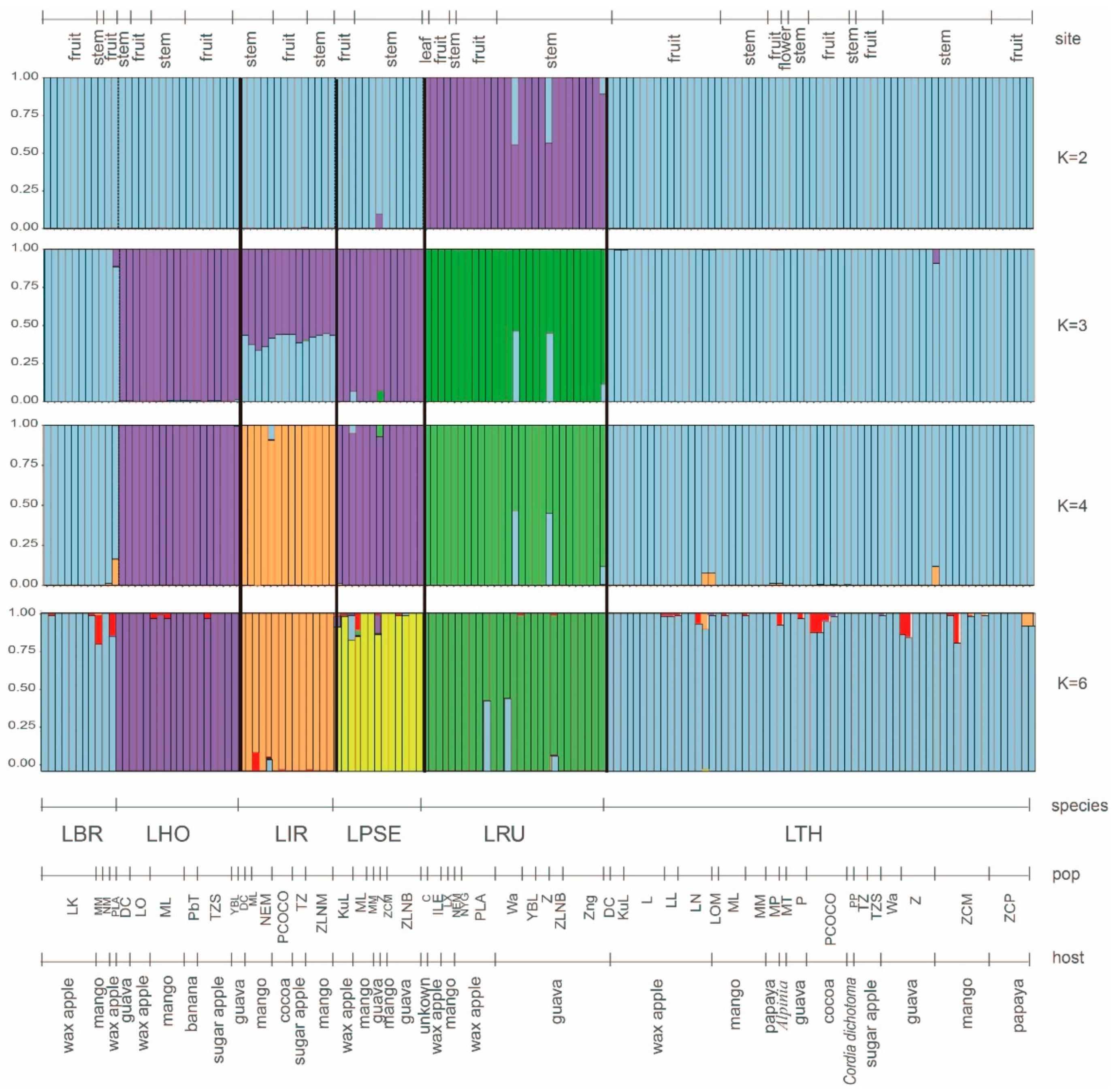
3.8. Genetic Differentiation and Genetic Structure in Lasiodiplodia Species
4. Discussion
4.1. Genetic Differentiation and Genetic Structure in Lasiodiplodia Species
4.2. Phylogenetic Relationships and Species Delimitation within the Lasiodiplodia Genus
4.3. Signs of Population Expansion, Subdivision, and Cryptic Sex in Lasiodiplodia Species
4.4. Intraspecies Variations and Host-Associated Genetic Differentiation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Punithalingam, E. Plant diseases attributed to Botryodiplodia theobromae Pat. In Biblioteca Mycologica; J. Cramer: Berlin, Germany, 1980; p. 113. [Google Scholar]
- MYCOBANK Database. Available online: http://www.mycobank.org (accessed on 18 June 2023).
- Correia, K.C.; Silva, M.A.; de Morais, M.A., Jr.; Armengol, J.; Phillips, A.J.; Cômara, M.; Michereff, S.J. Phylogeny, distribution and pathogenicity of Lasiodiplodia species associated with dieback of table grape in the main Brazilian exporting region. Plant Pathol. 2016, 65, 92–103. [Google Scholar] [CrossRef]
- Savant, N.V.; Raut, S.P. Studies on symptomatology of die-back of mango stone grafts. Acta Hort. 2000, 509, 823–832. [Google Scholar] [CrossRef]
- Sharma, I. A note on population dynamics and etiology of die back of mango in Himachal Pradesh. New Agricult. 1993, 2, 229–230. [Google Scholar]
- Ali, S.S.; Asman, A.; Shao, J.; Balidion, J.F.; Strem, M.D.; Puig, A.S.; Meinhardt, L.W.; Bailey, B.A. Genome and transcriptome analysis of the latent pathogen Lasiodiplodia theobromae, an emerging threat to the cacao industry. Genome 2020, 63, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.D.O.; Michereff, S.J.; Martins, R.B.; Gava, C.T.; Mizubuti, E.S.G.; Câmara, M.P.S. Species of Botryosphaeriaceae associated on mango in Brazil. Eur. J. Plant Pathol. 2010, 127, 509–519. [Google Scholar] [CrossRef]
- Phillips, A.J.L.; Alves, A.; Abdollahzadeh, J.; Slippers, B.; Wingfield, M.J.; Groenewald, J.Z.; Crous, P.W. The Botryosphaeriaceae: Genera and species known from culture. Stud. Mycol. 2013, 76, 51–167. [Google Scholar] [CrossRef]
- de Silva, N.I.; Phillips, A.J.; Liu, J.-K.; Lumyong, S.; Hyde, K.D. Phylogeny and morphology of Lasiodiplodia species associated with Magnolia forest plants. Sci. Rep. 2019, 9, 14355. [Google Scholar] [CrossRef]
- Dou, Z.P.; He, W.; Zhang, Y. Lasiodiplodia chinensis, a new holomorphic species from China. Mycosphere 2017, 8, 521–532. [Google Scholar] [CrossRef]
- Tennakoon, D.S.; Phillips, A.J.L.; Phookamsak, R.; Ariyawansa, H.A.; Bahkali, A.H.; Hyde, K.D. Sexual morph of Lasiodiplodia pseudotheobromae (Botryosphaeriaceae, Botryosphaeriales, Dothideomycetes) from China. Mycosphere 2016, 7, 990–1000. [Google Scholar] [CrossRef]
- Trakunyingcharoen, T.; Lombard, L.; Groenewald, J.Z.; Cheewangkoon, R.; To-Anun, C.; Crous, P.W. Caulicolous Botryosphaeriales from Thailand. Pers. Mol. Phylogeny Evol. 2015, 34, 87–99. [Google Scholar] [CrossRef]
- Liu, J.K.; Phookamsak, R.; Doilom, M.; Wikee, S.; Li, Y.M.; Ariyawansha, H.; Boonmee, S.; Chomnunti, P.; Dai, D.Q.; Bhat, J.D. Towards a natural classification of Botryosphaeriales. Fungal Divers. 2012, 57, 149–210. [Google Scholar] [CrossRef]
- Wingfield, M.J.; De Beer, Z.W.; Slippers, B.; Wingfield, B.D.; Groenewald, J.Z.; Lombard, L.; Crous, P.W. One fungus, one name promotes progressive plant pathology. Mol. Plant Pathol. 2012, 13, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.D.; Verma, K.S.; Singh, K.; Kaur, R. Morphological, pathological and molecular variability in Botryodiplodia theobromae (Botryosphaeriaceae) isolates associated with die-back and bark canker of pear trees in Punjab, India. Genet. Mol. Res. 2010, 9, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Munirah, M.; Azmi, A.; Yong, S.; Nur Ain Izzati, M. Characterization of Lasiodiplodia theobromae and L. pseudotheobromae causing fruit rot on pre-harvest mango in Malaysia. Plant Pathol. Quar. 2017, 7, 202–213. [Google Scholar] [CrossRef]
- Urbez-Torres, J.R.; Peduto, F.; Striegler, R.; Urrea-Romero, K.; Rupe, J.; Cartwright, R.; Gubler, W. Characterization of fungal pathogens associated with grapevine trunk diseases in Arkansas and Missouri. Fungal Dive. 2012, 52, 169–189. [Google Scholar] [CrossRef]
- Abdollahzadeh, J.; Javadi, A.; Goltapeh, E.M.; Zare, R.; Phillips, A.J.L. Phylogeny and morphology of four new species of Lasiodiplodia from Iran. Pers. Mol. Phylogeny Evol. 2010, 25, 1–10. [Google Scholar] [CrossRef]
- Ismail, A.M.; Cirvilleri, G.; Polizzi, G.; Crous, P.W.; Groenewald, J.Z.; Lombard, L. Lasiodiplodia species associated with dieback disease of mango (Mangifera indica) in Egypt. Australas. Plant Pathol. 2012, 41, 649–660. [Google Scholar] [CrossRef]
- Marques, M.W.; Lima, N.B.; de Morais, M.A.; Barbosa, M.A.G.; Souza, B.O.; Michereff, S.J.; Phillips, A.J.; Câmara, M.P. Species of Lasiodiplodia associated with mango in Brazil. Fungal Divers. 2013, 61, 181–193. [Google Scholar] [CrossRef]
- Sakalidis, M.L.; Ray, J.D.; Lanoiselet, V.; Hardy, G.E.S.; Burgess, T.I. Pathogenic Botryosphaeriaceae associated with Mangifera indica in the Kimberley region of Western Australia. Eur. J. Plant Pathol. 2011, 130, 379–391. [Google Scholar] [CrossRef]
- Damm, U.; Crous, P.W.; Fourie, P.H. Botryosphaeriaceae as potential pathogens of Prunus species in South Africa, with descriptions of Diplodia africana and Lasiodiplodia plurivora sp. nov. Mycologia 2007, 99, 664–680. [Google Scholar] [CrossRef]
- Machado, A.R.; Pinho, D.B.; Pereira, O.L. Phylogeny, identification and pathogenicity of the Botryosphaeriaceae associated with collar and root rot of the biofuel plant Jatropha curca s in Brazil, with a description of new species of Lasiodiplodia. Fungal Divers. 2014, 67, 231–247. [Google Scholar] [CrossRef]
- Crous, P.W.; Slippers, B.; Wingfield, M.J.; Rheeder, J.; Marasas, W.F.; Philips, A.J.; Alves, A.; Burgess, T.; Barber, P.; Groenewald, J.Z. Phylogenetic lineages in the Botryosphaeriaceae. Stud. Mycol. 2006, 55, 235–253. [Google Scholar] [CrossRef] [PubMed]
- De Wet, J.; Slippers, B.; Preisig, O.; Wingfield, B.D.; Wingfield, M.J. Phylogeny of the Botryosphaeriaceae reveals patterns of host association. Mol. Phylogenet. Evol. 2008, 46, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Gálvez, E.; Guerrero, P.; Barradas, C.; Crous, P.W.; Alves, A. Phylogeny and pathogenicity of Lasiodiplodia species associated with dieback of mango in Peru. Fungal Biol. 2017, 121, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.Y.; Xie, Y.; Zhang, W.; Wang, Y.; Liu, J.K.; Hyde, K.D.; Seem, R.C.; Zhang, G.Z.; Wang, Z.Y.; Yao, S.W. Species of Botryosphaeriaceae involved in grapevine dieback in China. Fungal Divers. 2013, 61, 221–236. [Google Scholar] [CrossRef]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 14. [Google Scholar] [CrossRef]
- Lücking, R.; Hawksworth, D. Formal description of sequence-based voucherless Fungi: Promises and pitfalls, and how to resolve them. IMA Fungus 2018, 9, 143–165. [Google Scholar] [CrossRef] [PubMed]
- Cruywagen, E.M.; Slippers, B.; Roux, J.; Wingfield, M.J. Phylogenetic species recognition and hybridization in Lasiodiplodia: A case study on species from baobabs. Fungal Biol. 2017, 121, 420–436. [Google Scholar] [CrossRef] [PubMed]
- Hawksworth, D.L.; Lücking, R. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol. Spectr. 2017, 5, 10-1128. [Google Scholar] [CrossRef]
- Slippers, B.; Wingfield, M.J. Botryosphaeriaceae as endophytes and latent pathogens of woody plants: Diversity, ecology and impact. Fungal Biol. Rev. 2007, 21, 90–106. [Google Scholar] [CrossRef]
- Yang, T.; Groenewald, J.Z.; Cheewangkoon, R.; Jami, F.; Abdollahzadeh, J.; Lombard, L.; Crous, P.W. Families, genera, and species of Botryosphaeriales. Fungal Biol. 2016, 121, 322–346. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.L.; Hsieh, H.Y. Occurrence and pathogenicity of stem canker of guava in Taiwan caused by Botryosphaeria rhodina. Plant Pathol. Bull. 2006, 15, 219–230. [Google Scholar]
- Ni, H.F.; Yang, H.R.; Chen, R.S.; Liou, R.F.; Hung, T.H. New Botryosphaeriaceae fruit rot of mango in Taiwan: Identification and pathogenicity. Bot. Stud. 2012, 53, 467–478. [Google Scholar]
- Wang, H.L.; Chen, P.H.; Ni, H.F.; Chen, R.S. Physiological characterization and screen of control chemicals for Lasiodiplodia theobromae of papaya. Plant Pathol. Bull. 2007, 16, 71–77. [Google Scholar]
- Ni, H.F.; Chuang, M.F.; Hsu, S.L.; Lai, S.Y.; Yang, H.R. Survey of Botryosphaeria spp., causal agents of postharvest disease of avocado, in Taiwan. J. Taiwan Agric. Res. 2011, 60, 157–166. [Google Scholar]
- Ni, H.F.; Chen, R.S.; Chang, S.F.; Yang, H.R. First report of Lasiodiplodia fruit rot of jackfruit in Taiwan. Plant Dis. 2008, 92, 1137. [Google Scholar] [CrossRef]
- Ko, W.H.; Wang, I.T.; Ann, P.J. Lasiodiplodia theobromae as a causal agent of kumquat dieback in Taiwan. Plant Dis. 2004, 88, 1383. [Google Scholar] [CrossRef]
- Patrice, N.D.J.; Alain, H.; Nourou, K.N.A.; Bertrand, M.S.; Christine, A.D.B.; Brice, T.T.D.; Zachee, A. Dieback of cashew (Anacardium occidentale) in Cameroon. Pakistan J. Phytopathol. 2020, 32, 231–239. [Google Scholar] [CrossRef]
- Huang, C.; Chang, Y.; Chao, J.; Huang, J. Fruit rot of passion fruit caused by Lasiodiplodia theobromae in Taiwan. J. Taiwan Agric. Res. 2019, 68, 333–340. [Google Scholar]
- Guo, Z. Stem blight of mung bean caused by Botryodiplodia theobromae. Plant Prot. Bull. 1998, 40, 315–327. [Google Scholar]
- Su, J.F.; Ann, P.J.; Yang, C.W.; Hsu, Z.H.; Chern, L.L.; Tsay, H.C.; Huang, C.C.; Hsieh, T.F. Black rot of braided seedlings of Malabar chestnut caused by Lasiodiplodia theobromae in Taiwan. Plant Pathol. Bull. 2014, 23, 263–269. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017; Available online: https://www.R-project.org (accessed on 18 June 2023).
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant Graphics for Data Analysis (2nd ed.). Meas. Interdiscip. Res. Perspect. 2019, 17, 160–167. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. A Guide Methods Appl. 1990, 18, 315–322. [Google Scholar]
- Phillips, A.; Alves, A.; Correia, A.; Luque, J. Two new species of Botryosphaeria with brown, 1-septate ascospores and Dothiorella anamorphs. Mycologia 2005, 97, 513–529. [Google Scholar] [CrossRef]
- Glass, N.L.; Donaldson, G.C. Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microbiol. 1995, 61, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods. 2012, 9, 772. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Felsenstein, J. PHYLIP (Phylogeny Inference Package) Version 3.6. Distributed by the Author; Department of Genome Sciences, University of Washington: Seattle, WA, USA, 2005. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree v1.4.4. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 18 June 2023).
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble species by automatic partitioning. Mol. Ecol. Resour. 2021, 21, 609–620. [Google Scholar] [CrossRef]
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Colombia University Press: New York, NY, USA, 1987. [Google Scholar]
- Hudson, R.R.; Kaplan, N.L. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 1985, 111, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.K. A test of neutrality based on interlocus associations. Genetics 1997, 146, 1197–1206. [Google Scholar] [CrossRef]
- Hill, W.G.; Robertson, A. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 1968, 38, 226–231. [Google Scholar] [CrossRef]
- Pennings, P.S.; Hermisson, J. Soft sweeps III: The signature of positive selection from recurrent mutation. PLoS Genet. 2006, 2, e186. [Google Scholar] [CrossRef]
- Rozas, J.; Gullaud, M.; Blandin, G.; Aguadé, M. DNA variation at the rp49 gene region of Drosophila simulans: Evolutionary inferences from an unusual haplotype structure. Genetics 2001, 158, 1147–1155. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.-X.; Li, W.-H. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Excoffier, L. Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: Application to human mitochondrial DNA. Genetics 1999, 152, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Harpending, H.C. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum. Biol. 1994, 66, 591–600. [Google Scholar] [PubMed]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar]
- Ray, N.; Currat, M.; Excoffier, L. Intra-deme molecular diversity in spatially expanding populations. Mol. Biol. Evol. 2003, 20, 76–86. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Wright, S. Genetical structure of populations. Nature 1950, 166, 247–249. [Google Scholar] [CrossRef]
- Taylor, J.W.; Geiser, D.M.; Burt, A.; Koufopanou, V. The evolutionary biology and population genetics underlying fungal strain typing. Clin. Microbiol. Rev. 1999, 12, 126–146. [Google Scholar] [CrossRef]
- Wright, S. Evolution in mendelian populations. Genetics 1931, 16, 97–159. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; VonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.M. pophelper: An R package and web app to analyze and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef]
- Ni, H.F.; Liou, R.F.; Hung, T.H.; Chen, R.S.; Yang, H.R. First report of fruit rot disease of mango caused by Botryosphaeria dothidea and Neofusicoccum mangiferae in Taiwan. Plant Dis. 2010, 94, 128. [Google Scholar] [CrossRef]
- Slippers, B.; Johnson, G.I.; Crous, P.W.; Coutinho, T.A.; Wingfield, B.D.; Wingfield, M.J. Phylogenetic and morphological re-evaluation of the Botryosphaeria species causing diseases of Mangifera indica. Mycologia 2005, 97, 99–110. [Google Scholar] [CrossRef]
- Alves, A.; Crous, P.W.; Correia, A.; Phillips, A. Morphological and molecular data reveal cryptic speciation in Lasiodiplodia theobromae. Fungal Divers. 2008, 28, 1–13. [Google Scholar]
- Santos, P.H.D.; Carvalho, B.M.; Aguiar, K.P.; Aredes, F.A.S.; Poltronieri, T.P.S.; Vivas, J.M.S.; Dias, V.M.; Bezerra, G.A.; Pinho, D.B.; Pereira, M.G. Phylogeography and population structure analysis reveals diversity by mutations in Lasiodiplodia theobromae with distinct sources of selection. Genet. Mol. Res. 2017, 16, 1–14. [Google Scholar] [CrossRef]
- Grant, W.A.S.; Bowen, B.W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. J. Hered. 1998, 89, 415–426. [Google Scholar] [CrossRef]
- Hobbs, J.-P.A.; Van Herwerden, L.; Jerry, D.R.; Jones, G.P.; Munday, P.L. High genetic diversity in geographically remote populations of endemic and widespread coral reef angelfishes (genus: Centropyge). Diversity 2013, 5, 39–50. [Google Scholar] [CrossRef]
- Slippers, B.; Crous, P.W.; Jami, F.; Groenewald, J.Z.; Wingfield, M.J. Diversity in the Botryosphaeriales: Looking back, looking forward. Fungal Biol. 2017, 121, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, S.; Zhao, L.; Sun, X.; He, W.; Zhang, Y.; Dai, Y.-C. Lasiodiplodia spp. associated with Aquilaria crassna in Laos. Mycol. Prog. 2019, 18, 683–701. [Google Scholar] [CrossRef]
- Custódio, F.A.; Machado, A.R.; Soares, D.J.; Pereira, O.L. Lasiodiplodia hormozganensis causing basal stem rot on Ricinus communis in Brazil. Australas. Plant Dis. Notes 2018, 13, 25. [Google Scholar] [CrossRef]
- Coutinho, I.B.L.; Freire, F.C.O.; Lima, C.S.; Lima, J.S.; Gonçalves, F.J.T.; Machado, A.R.; Silva, A.M.S.; Cardoso, J.E. Diversity of genus Lasiodiplodia associated with perennial tropical fruit plants in northeastern Brazil. Plant Pathol. 2017, 66, 90–104. [Google Scholar] [CrossRef]
- Dissanayake, A.J.; Phillips, A.J.L.; Li, X.H.; Hyde, K.D. Botryosphaeriaceae: Current status of genera and species. Mycosphere 2016, 7, 1001–1073. [Google Scholar] [CrossRef]
- Hyde, K.D.; Tennakoon, D.S.; Jeewon, R.; Bhat, D.J.; Maharachchikumbura, S.S.N.; Rossi, W.; Leonardi, M.; Lee, H.B.; Mun, H.Y.; Houbraken, J. Fungal diversity notes 1036–1150: Taxonomic and phylogenetic contributions on genera and species of fungal taxa. Fungal Divers. 2019, 96, 1–242. [Google Scholar] [CrossRef]
- Jayasiri, S.C.; Hyde, K.D.; Jones, E.B.G.; McKenzie, E.H.C.; Jeewon, R.; Phillips, A.J.L.; Bhat, D.J.; Wanasinghe, D.N.; Liu, J.K.; Lu, Y.Z. Diversity, morphology and molecular phylogeny of Dothideomycetes on decaying wild seed pods and fruits. Mycosphere 2019, 10, 1–186. [Google Scholar] [CrossRef]
- Meng, C.R.; Zhang, Q.; Yang, Z.F.; Geng, K.; Zeng, X.Y.; Chethana, K.T.; Wang, Y. Lasiodiplodia syzygii sp. nov.(Botryosphaeriaceae) causing post-harvest water-soaked brown lesions on Syzygium samarangense in Chiang Rai, Thailand. Biodivers. Data J. 2021, 9, e60604. [Google Scholar] [CrossRef]
- Zhang, W.; Groenewald, J.Z.; Lombard, L.; Schumacher, R.K.; Phillips, A.J.L.; Crous, P.W. Evaluating species in Botryosphaeriales. Pers. Mol. Phylogeny Evol. Fungi 2021, 46, 63–115. [Google Scholar] [CrossRef]
- Trakunyingcharoen, T.; Cheewangkoon, R.; To-anun, C. Phylogeny and pathogenicity of fungal species in the family Botryosphaeriaceae associated with mango (Mangifera indica) in Thailand. Int. J. Agric. Technol. 2013, 9, 1535–1543. [Google Scholar]
- van der Linde, J.A.; Six, D.L.; Wingfield, M.J.; Roux, J. Lasiodiplodia species associated with dying Euphorbia ingens in South Africa. South. For. 2011, 73, 165–173. [Google Scholar] [CrossRef]
- Kubatko, L.S.; Degnan, J.H. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 2007, 56, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C.; Shapira, J.F.; Daniel, S.W.; Aquadro, C.F.; Lansman, R.A. Mitochondrial DNA differentiation during the speciation process in Peromyscus. Mol. Biol. Evol. 1983, 1, 38–56. [Google Scholar] [PubMed]
- Maddison, W.P.; Knowles, L.L. Inferring phylogeny despite incomplete lineage sorting. Syst. Biol. 2006, 55, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Pamilo, P.; Nei, M. Relationships between gene trees and species trees. Mol. Biol. Evol. 1988, 5, 568–583. [Google Scholar]
- Rosenberg, N.A. The probability of topological concordance of gene trees and species trees. Theor. Popul. Biol. 2002, 61, 225–247. [Google Scholar] [CrossRef] [PubMed]
- Haelewaters, D.; De Kesel, A.; Pfister, D.H. Integrative taxonomy reveals hidden species within a common fungal parasite of ladybirds. Sci. Rep. 2018, 8, 15966. [Google Scholar] [CrossRef]
- Netto, M.S.B.; Assunção, I.P.; Lima, G.S.A.; Marques, M.W.; Lima, W.G.; Monteiro, J.H.A.; de Queiroz Balbino, V.; Michereff, S.J.; Phillips, A.J.L.; Câmara, M.P.S. Species of Lasiodiplodia associated with papaya stem-end rot in Brazil. Fungal Divers. 2014, 67, 127–141. [Google Scholar] [CrossRef]
- Netto, M.S.B.; Lima, W.G.; Correia, K.C.; Da Silva, C.F.B.; Thon, M.; Martins, R.B.; Miller, R.N.G.; Michereff, S.J.; Câmara, M.P.S. Analysis of phylogeny, distribution, and pathogenicity of Botryosphaeriaceae species associated with gummosis of Anacardium in Brazil, with a new species of Lasiodiplodia. Fungal Biol. 2017, 121, 437–451. [Google Scholar] [CrossRef]
- Burt, A.; Carter, D.A.; Koenig, G.L.; White, T.J.; Taylor, J.W. Molecular markers reveal cryptic sex in the human pathogen Coccidioides immitis. Proc. Natl. Acad. Sci. USA 1996, 93, 770–773. [Google Scholar] [CrossRef]
- Lee, S.C.; Ni, M.; Li, W.; Shertz, C.; Heitman, J. The evolution of sex: A perspective from the fungal kingdom. Microbiol. Mol. Biol. Rev. 2010, 74, 298–340. [Google Scholar] [CrossRef]
- Schurko, A.M.; Neiman, M.; Logsdon, J.M. Signs of sex: What we know and how we know it. Trends. Ecol. Evol. 2009, 24, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Billiard, S.; López-Villavicencio, M.; Hood, M.E.; Giraud, T. Sex, outcrossing and mating types: Unsolved questions in fungi and beyond. J. Evol. Biol. 2012, 25, 1020–1038. [Google Scholar] [CrossRef] [PubMed]
- Boyd, O.F.; Philips, T.K.; Johnson, J.R.; Nixon, J.J. Geographically structured genetic diversity in the cave beetle Darlingtonea kentuckensis Valentine, 1952 (Coleoptera, Carabidae, Trechini, Trechina). Subterr. Biol. 2020, 34, 1–23. [Google Scholar] [CrossRef]
- Burgess, T.I.; Barber, P.A.; Mohali, S.; Pegg, G.; de Beer, W.; Wingfield, M.J. Three new Lasiodiplodia spp. from the tropics, recognized based on DNA sequence comparisons and morphology. Mycologia 2006, 98, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Linaldeddu, B.T.; Deidda, A.; Scanu, B.; Franceschini, A.; Serra, S.; Berraf-Tebbal, A.; Zouaoui Boutiti, M.; Ben Jamâa, M.L.; Phillips, A.J.L. Diversity of Botryosphaeriaceae species associated with grapevine and other woody hosts in Italy, Algeria and Tunisia, with descriptions of Lasiodiplodia exigua and Lasiodiplodia mediterranea sp. nov. Fungal Divers. 2015, 71, 201–214. [Google Scholar] [CrossRef]
- Pavlic, D.; Slippers, B.; Coutinho, T.A.; Gryzenhout, M.; Wingfield, M.J. Lasiodiplodia gonubiensis sp. nov., a new Botryosphaeria anamorph from native Syzygium cordatum in South Africa. Stud. Myco. 2004, 50, 313–322. [Google Scholar]
- Pavlic, D.; Wingfield, M.J.; Barber, P.; Slippers, B.; Hardy, G.E.S.J.; Burgess, T.I. Seven new species of the Botryosphaeriaceae from baobab and other native trees in Western Australia. Mycologia 2008, 100, 851–866. [Google Scholar] [CrossRef]
- Begoude, B.A.D.; Slippers, B.; Wingfield, M.J.; Roux, J. Botryosphaeriaceae associated with Terminalia catappa in cameroon, south africa and madagascar. Mycol. Prog. 2010, 9, 101–123. [Google Scholar] [CrossRef]
- Kawecki, T.J. Red queen meets Santa Rosalia: Arms races and the evolution of host specialization in organisms with parasitic lifestyles. Am. Nat. 1998, 152, 635–651. [Google Scholar] [CrossRef]
- Giraud, T.; Enjalbert, J.; Fournier, E.; Delmotte, F.; Dutech, C. Population genetics of fungal diseases of plants. Parasite 2008, 15, 449–454. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | Host | No. of Isolates (No. of Locations) |
|---|---|---|
| Lasiodiplodia theobromae | Syzygium samarangense (wax apple) | 15 (4) |
| Psidium guajava (guava) | 12 (4) | |
| Mangifera indica (mango) | 16 (4) | |
| Carica papaya (papaya) | 8 (2) | |
| Annona squamosa (sugar apple) | 4 (2) | |
| Theobroma cacao (cocoa) | 6 (1) | |
| Cordia dichotoma | 1 (1) | |
| Alpinia | 1 (1) | |
| Lasiodiplodia brasiliensis | Syzygium samarangense (wax apple) | 9 (2) |
| Mangifera indica (mango) | 2 (2) | |
| Lasiodiplodia hormozganensis | Syzygium samarangense (wax apple) | 3 (1) |
| Mangifera indica (mango) | 5 (1) | |
| Psidium guajava (guava) | 3 (2) | |
| Annona squamosa (sugar apple) | 5 (1) | |
| Musa spp. (banana) | 2 (1) | |
| Lasiodiplodia pseudotheobromae | Syzygium samarangense (wax apple) | 3 (1) |
| Psidium guajava (guava) | 6 (2) | |
| Mangifera indicnga (mango) | 4 (3) | |
| Lasiodiplodia rubropurpurea | Syzygium samarangense (wax apple) | 8 (3) |
| Psidium guajava (guava) | 16 (5) | |
| Mangifera indica (mango) | 2 (2) | |
| Lasiodiplodia iranensis | Mangifera indica (mango) | 8 (3) |
| Psidium guajava (guava) | 1 (1) | |
| Annona squamosa (sugar apple) | 2 (1) | |
| Theobroma cacao (cocoa) | 3 (1) | |
| Total | 145 |
| S | h | Hd | π (×10−3) | ZnS | ZZ | RM | Fu and Li’s D * | Tajima’s D | |
|---|---|---|---|---|---|---|---|---|---|
| L. theobromae | |||||||||
| ITS | 2 | 3 | 0.12 | 0.24 | 0.00 | 0.00 | 0 | 0.72 | −1.19 |
| SSU | 18 | 11 | 0.29 | 0.66 | 0.11 | 0.22 | 0 | −4.76 * | −2.51 ** |
| EF1 | 31 | 12 | 0.70 | 3.99 | 0.17 | 0.13 | 2 | −1.22 | −2.08 * |
| TUB | 6 | 6 | 0.24 | 0.70 | 0.07 | 0.13 | 0 | −0.63 | −1.84 * |
| Combined dataset | 57 | 27 | 0.87 | 1.36 | 0.07 | 0.24 | 3 | −2.66 * | −2.41 ** |
| L. brasiliensis | |||||||||
| ITS | 6 | 6 | 0.80 | 3.73 | 0.54 | 0.12 | 1 | −0.44 | −0.23 |
| SSU | 11 | 3 | 0.35 | 1.98 | 0.82 | 0.08 | 0 | −2.43 * | −2.01 * |
| EF1 | 6 | 2 | 0.18 | 1.85 | 1.00 | 0.00 | 0 | −2.21 * | −1.85 * |
| TUB | 2 | 2 | 0.18 | 0.81 | 1.00 | 0.00 | 0 | −1.66 | −1.43 |
| Combined dataset | 25 | 7 | 0.87 | 2.11 | 0.32 ** | 0.38 | 1 | −2.12 * | −1.70 |
| L. hormozganensis | |||||||||
| ITS | 6 | 6 | 0.86 | 3.46 | 0.13 | −0.06 | 0 | 1.26 | 0.09 |
| SSU | 6 | 6 | 0.49 | 2.30 | 0.15 | 0.14 | 0 | −3.03 * | −2.15 ** |
| EF1 | 2 | 2 | 0.11 | 0.38 | 1.00 | 0.00 | 0 | −1.99 | −1.51 |
| TUB | 6 | 5 | 0.66 | 3.06 | 0.17 | 0.15 | 0 | −0.10 | −0.70 |
| Combined dataset | 22 | 13 | 0.95 | 1.68 | 0.10 | 0.14 | 1 | −1.23 | −1.30 |
| L. pseudotheobromae | |||||||||
| ITS | 2 | 2 | 0.15 | 0.60 | ND | ND | 0 | −1.78 | −1.47 |
| SSU | 15 | 7 | 0.73 | 3.12 | 0.31 | 0.05 | 0 | −1.37 | −1.45 |
| EF1 | 15 | 7 | 0.73 | 3.12 | 0.31 | 0.05 | 0 | −1.37 | −1.45 |
| TUB | 3 | 2 | 0.15 | 1.03 | 1.00 | 0.00 | 0 | −2.02 | −1.65 |
| Combined dataset | 26 | 10 | 0.95 | 2.04 | 0.19 | 0.21 | 1 | −1.91 | −1.65 |
| L. rubropurpurea | |||||||||
| ITS | 1 | 2 | 0.14 | 0.28 | ND | ND | 0 | 0.61 | −0.73 |
| SSU | 13 | 6 | 0.40 | 1.21 | 0.48 | 0.22 | 1 | −3.20 * | −2.16 * |
| EF1 | 44 | 8 | 0.84 | 13.90 | 0.61 | 0.18 | 1 | 1.49 * | −1.12 |
| TUB | 2 | 3 | 0.15 | 0.33 | 0.00 ** | 0.00 | 0 | −2.22 | −1.51 |
| Combined dataset | 60 | 16 | 0.90 | 6.11 | 0.36 | 0.35 | 4 | 0.16 | −1.48 |
| L. iranensis | |||||||||
| ITS | − | 1 | − | − | − | − | − | − | − |
| SSU | 13 | 6 | 0.40 | 1.21 | 0.48 | 0.22 | 1 | −3.20 * | −2.16 * |
| EF1 | 18 | 5 | 0.67 | 9.59 | 0.37 | −0.04 | 0 | −0.99 | −0.61 |
| TUB | 8 | 9 | 0.91 | 6.58 | 0.22 | −0.05 | 1 | 0.02 | 0.17 |
| Combined dataset | 37 | 13 | 0.99 | 3.72 | 0.18 | 0.20 | 2 | −1.29 | −0.90 |
| Sum of Squares | d.f. | Variance Components | Percentage of Variation | Fixation Indices | |
|---|---|---|---|---|---|
| Among species | 2629.35 | 5 | 24.02 | 89.18 | FCT = 0.89 * |
| Among populations within species | 207.57 | 46 | 0.97 | 3.60 | FSC = 0.33 * |
| Within populations | 182.76 | 94 | 1.94 | 7.22 | FST = 0.93 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, Y.-Z.; Liyanage, W.K.; Shih, H.-C.; Tseng, M.-N.; Shiao, M.-S.; Chiang, Y.-C. Unveiling Cryptic Species Diversity and Genetic Variation of Lasiodiplodia (Botryosphaeriaceae, Botryosphaeriales) Infecting Fruit Crops in Taiwan. J. Fungi 2023, 9, 950. https://doi.org/10.3390/jof9090950
Ko Y-Z, Liyanage WK, Shih H-C, Tseng M-N, Shiao M-S, Chiang Y-C. Unveiling Cryptic Species Diversity and Genetic Variation of Lasiodiplodia (Botryosphaeriaceae, Botryosphaeriales) Infecting Fruit Crops in Taiwan. Journal of Fungi. 2023; 9(9):950. https://doi.org/10.3390/jof9090950
Chicago/Turabian StyleKo, Ya-Zhu, Wasantha Kumara Liyanage, Huei-Chuan Shih, Min-Nan Tseng, Meng-Shin Shiao, and Yu-Chung Chiang. 2023. "Unveiling Cryptic Species Diversity and Genetic Variation of Lasiodiplodia (Botryosphaeriaceae, Botryosphaeriales) Infecting Fruit Crops in Taiwan" Journal of Fungi 9, no. 9: 950. https://doi.org/10.3390/jof9090950