
Transcriptome and Metabolome Integration Reveals the Impact of Fungal Elicitors on Triterpene Accumulation in Sanghuangporus sanghuang

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Fungal Elicitor
2.2. Strains and Cultivation
2.3. Fungal Elicitor Elicitation Test
2.4. Sample Preparation and Analysis of Metabolism
2.4.1. Specimen Preparation
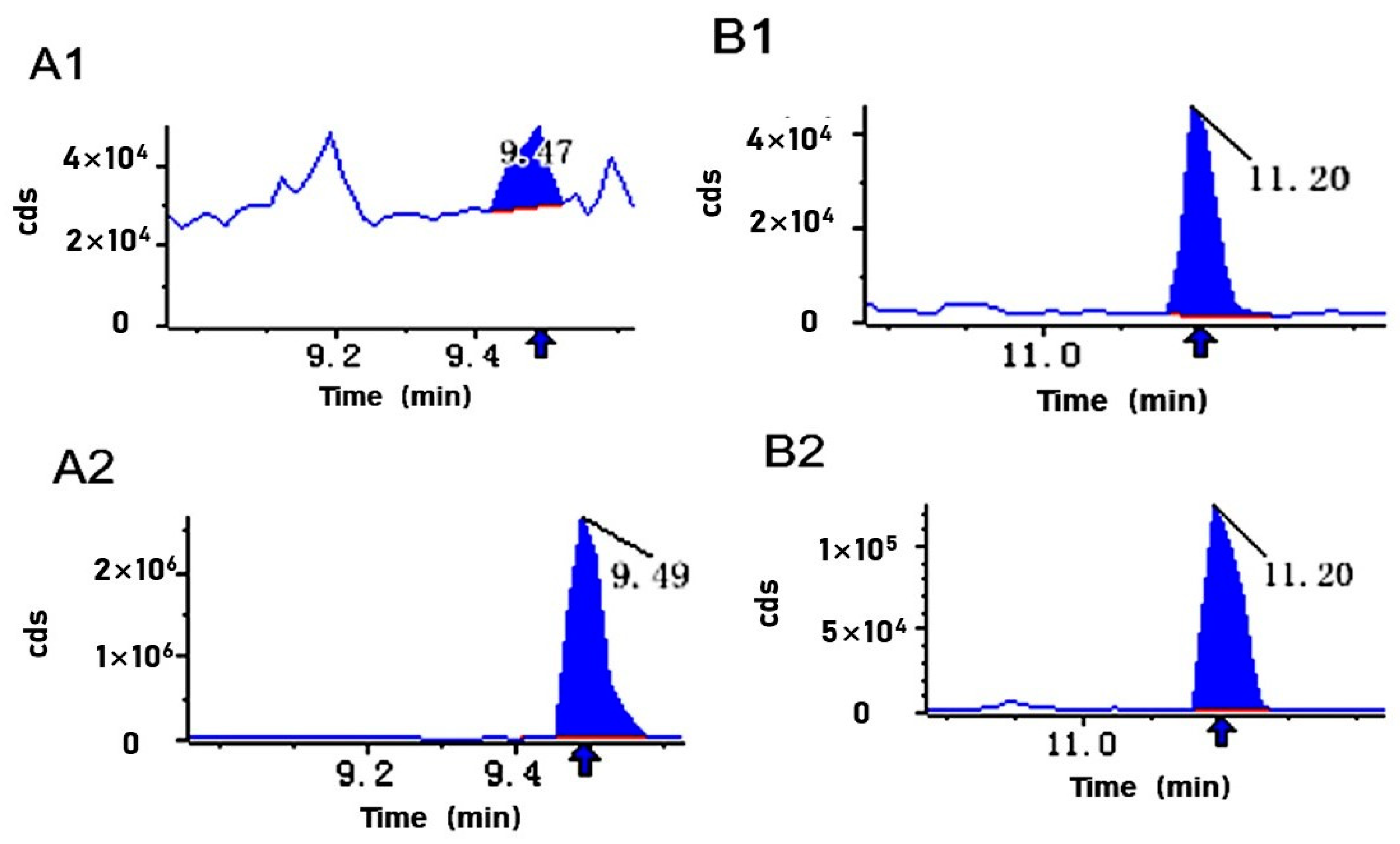
2.4.2. Liquid Chromatography–Mass Spectrometry Analysis
2.4.3. Metabolomic Data Analysis
2.4.4. RNA Extraction, cDNA Library Construction, and Sequencing
2.4.5. Transcriptome and Metabolomic Assembly and Annotation
2.4.6. Expression of Triterpenoid Biosynthesis-Related Genes in S. sanghuang under Elicitor Treatment
2.5. Determination of Total Triterpene Content
2.6. Detection of NO Content
2.7. Statistical Analysis
3. Results
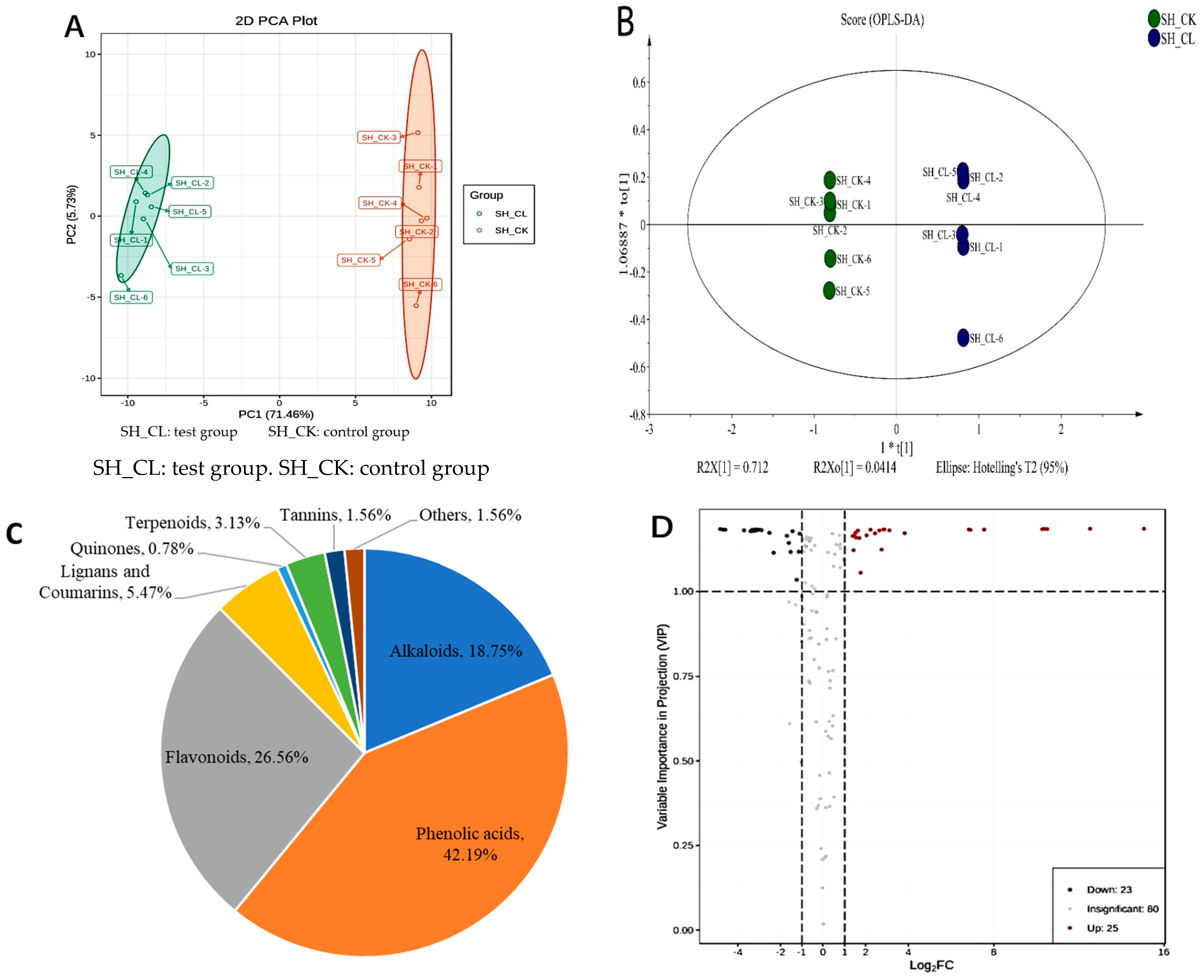
3.1. Identification of Metabolites
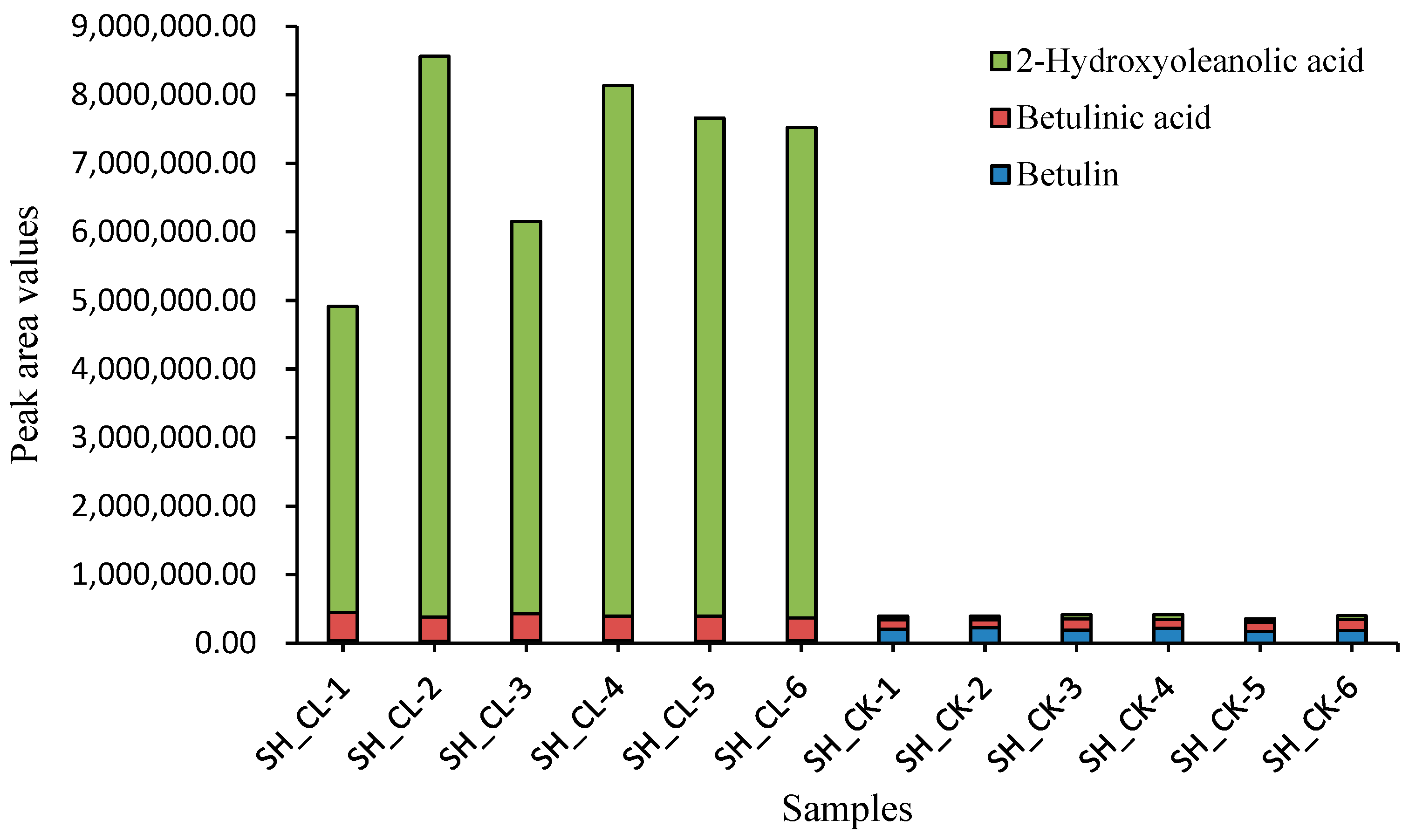
3.2. Variation in DAMs of Triterpenes
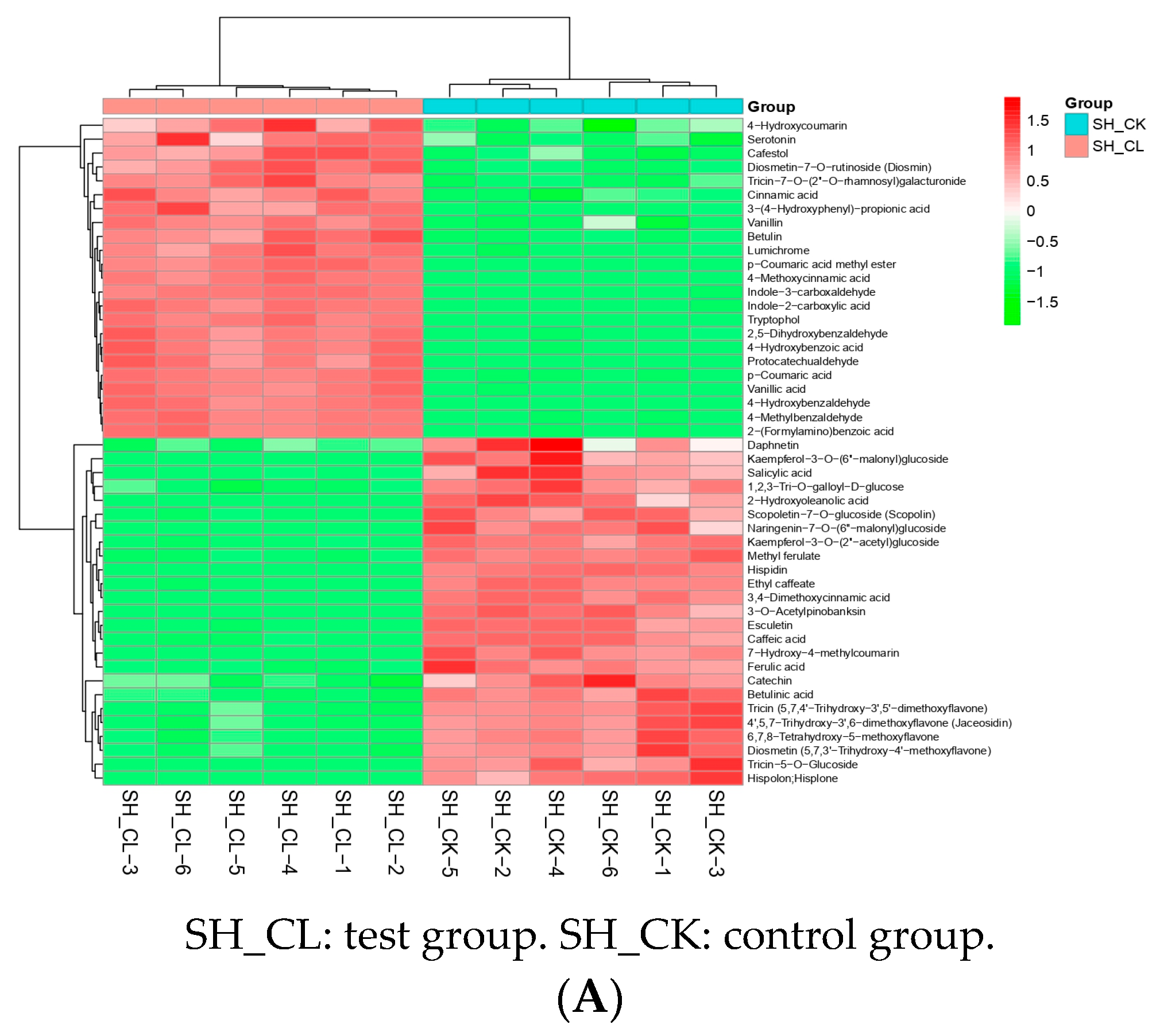
3.3. Filtering and Verification of Various Metabolites
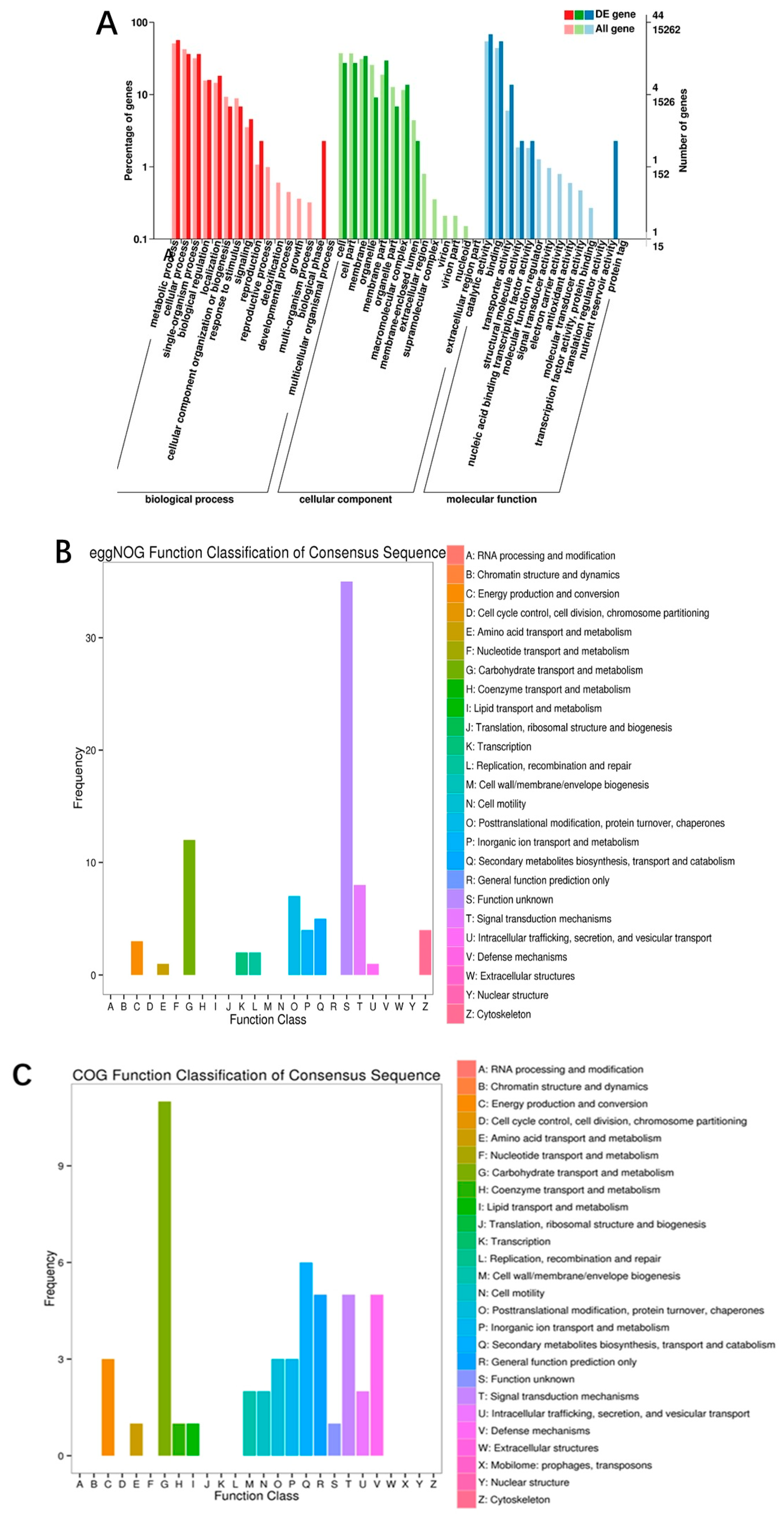
3.4. Transcriptomic Analysis of S. sanghuang from ET and WET
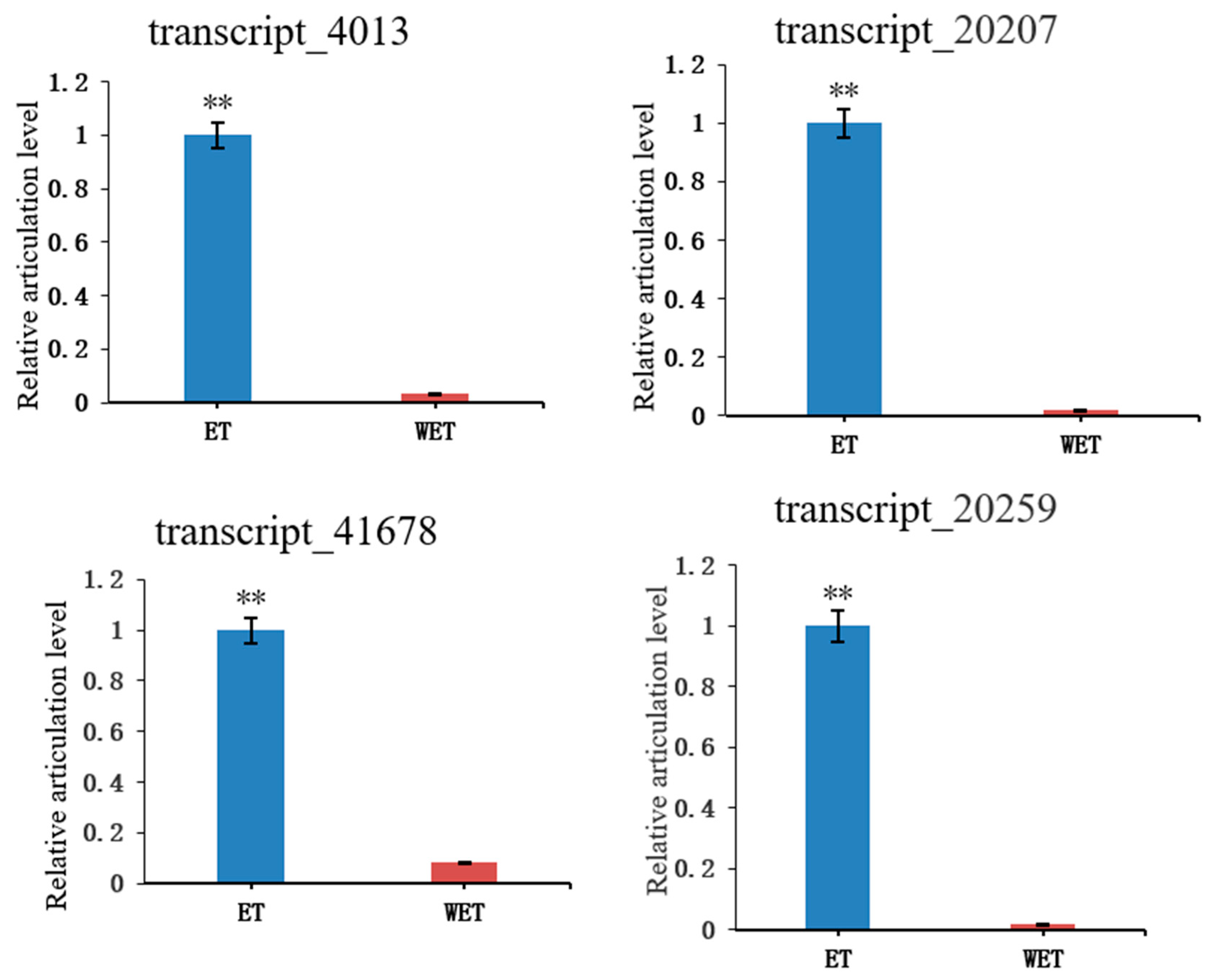
3.5. Verification of Differentially Expressed Transcripts (DETs) by qRT-PCR
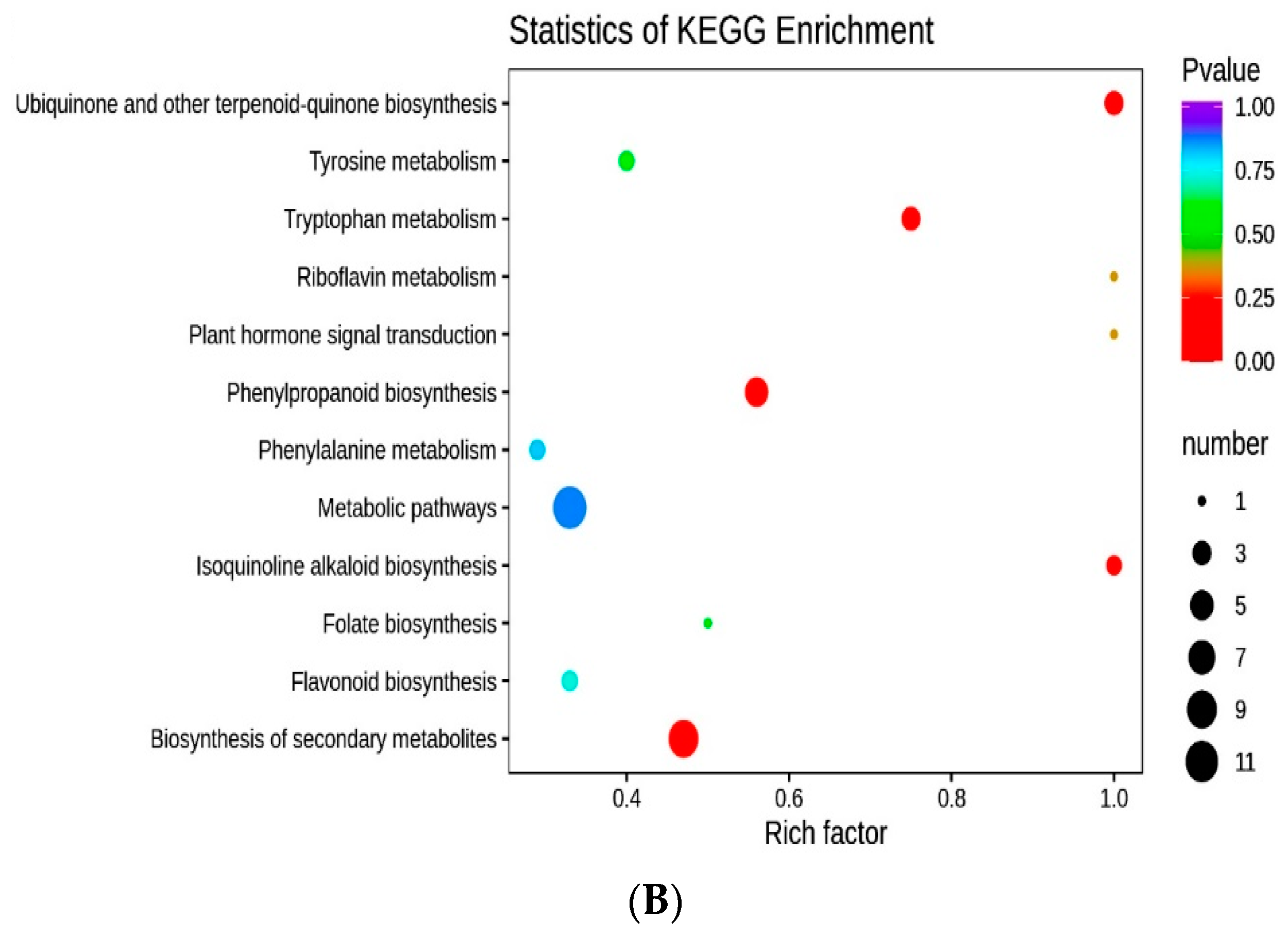
3.6. Correlation between DAMs and DETs
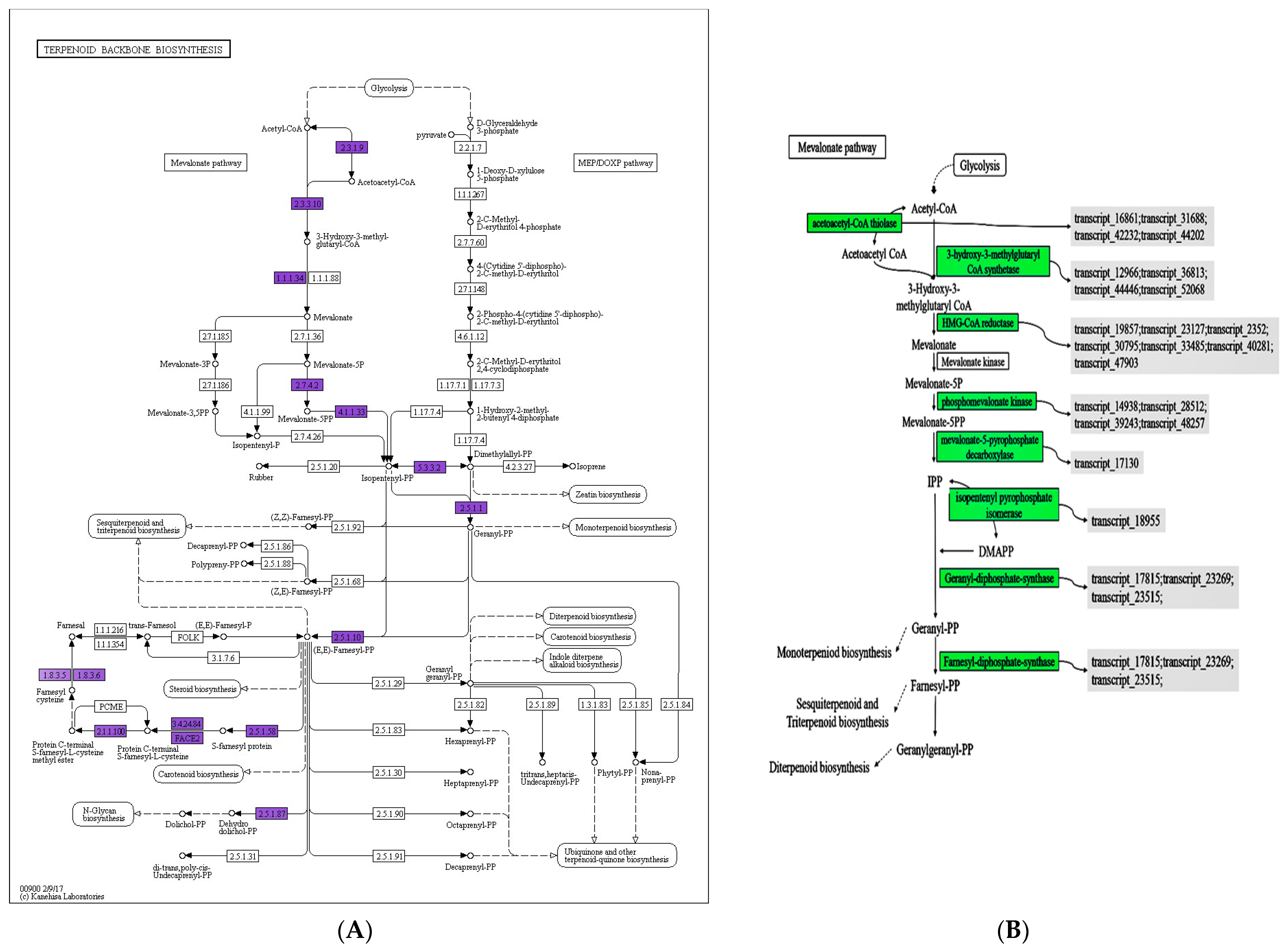
3.7. Effect of Mevalonate Pathways in ET Group
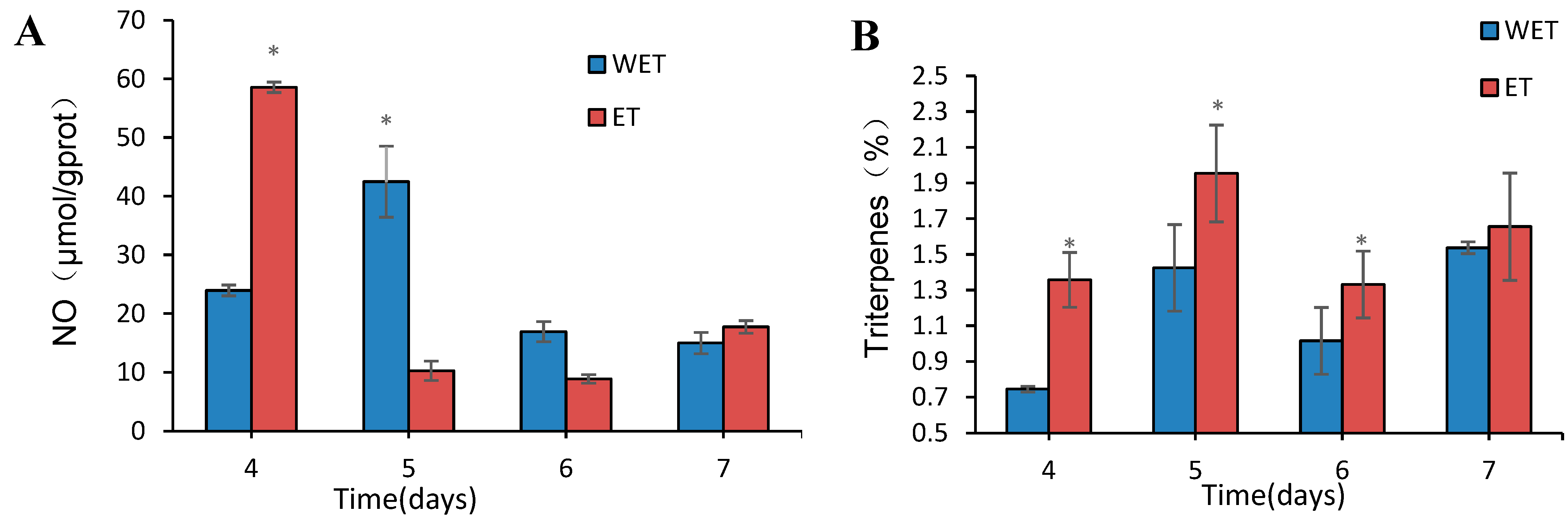
3.8. Detection of NO Content
3.9. Detection of Total Triterpene Content
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CC | correlation coefficient |
| CE | collision energy |
| CMCs | cambial meristematic cells |
| COGs | clusters of orthologous groups of proteins |
| CUR | curtain gas |
| DAMs | differentially accumulated metabolites |
| DEGs | differentially expressed genes |
| DETs | differentially expressed transcripts |
| DP | de-clustering potential |
| ESI | electrospray ionization |
| ET | elicitor-treated |
| GO | gene ontology |
| GPI-APs | glycosylphosphatidylinositol-anchored proteins |
| GSI | ion source gas I |
| GSII | ion source gas I |
| HPLC–MS/MS | high-performance liquid chromatography–tandem mass spectrometry |
| IS | ion spray voltage |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KOG | Eukaryotic Orthologous Groups |
| LIT | linear ion trap |
| MRM | multiple reaction monitoring |
| NEB | NEBNext® Ultra™ RNA library prep kit |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| Nr | non-redundant protein sequences |
| OPLS-DA | orthogonal partial least-squares discriminant analysis |
| PC1 | principal component 1 |
| PC2 | principal component 2 |
| PCA | principal component analysis |
| QQQ | triple quadrupole |
| QRT-PCR | quantitative real-time polymerase chain reaction |
| Q TRAP | triple quadrupole linear ion trap mass spectrometer |
| RNA-seq | RNA sequencing |
| S. sanghuang | Sanghuangporus sanghuang |
| SH-CL (ET) | elicitor-treated |
| SH-CK (WET) | without elicitor treatment |
| TIAs | terpenoid indole alkaloids |
| UPLC–MS/MS | ultra-performance liquid chromatography–mass spectrometry |
| UV | unit-variance scaling |
| VIP | variable importance in projection |
| WET | without elicitor treatment |
References
- Zhou, L.; GhobadNejhad, M.; Tian, X.; Wang, Y.; Wu, F. Current status of ‘Sanghuang’ as a group of medicinal mushrooms and their perspective in industry development. Food Rev. Int. 2022, 38, 589–607. [Google Scholar] [CrossRef]
- Wu, S.; Dai, Y.; Tsutomu, H.; Yu, T.; Wang, D.; Chang, H.; Shih, S. Species clarification for the medicinally valuable ‘sanghuang’ mushroom. Bot. Stud. 2012, 53, 135–149. Available online: https://ejournal.sinica.edu.tw/bbas/content/2012/1/Bot531-13.pdf (accessed on 12 December 2021).
- Zhou, L.; Vlasak, J.; Decock, C.; Assefa, A.; Stenlid, J.; Abate, D.; Wu, S.; Dai, Y. Global diversity and taxonomy of the Inonotus linteus complex (Hymenochaetales, Basidiomycota): Sanghuangporus gen. nov., Tropicoporus excentrodendri and T. guanacastensis gen. et spp. nov., and 17 new combinations. Fungal Divers. 2016, 77, 335–347. [Google Scholar] [CrossRef]
- Cheng, J.; Song, J.; Wei, H.; Wang, Y.; Huang, X.; Liu, Y.; Lu, N.; He, L.; Lv, G.; Ding, H.; et al. Structural characterization and hypoglycemic activity of an intracellular polysaccharide from Sanghuangporus sanghuang mycelia. Int. J. Biol. Macromol. 2020, 164, 3305–3314. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Ma, J.; Han, C.; Jin, Y.; Zhao, G.; He, X. Extraction and antioxidant activity of total triterpenoids in the mycelium of a medicinal fungus, Sanghuangporus sanghuang. Sci. Rep. 2019, 9, 7418. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, B.; Dai, H.; Ren, J.; Zhou, L.; Wu, S.; Liu, H. Sesquiterpenes and polyphenols with glucose-uptake stimulatory and antioxidant activities from the medicinal mushroom Sanghuangporus sanghuang. Chin. J. Nat. Med. 2021, 19, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Cai, C.; Liu, J.; Gao, S.; Zhao, G.; He, X. In vitro antibacterial and antitumor activity of total triterpenoids from a medicinal mushroom Sanghuangporus sanghuang (Agaricomycetes) in liquid fermentation culture. Int. J. Med. Mushrooms 2021, 23, 27–39. [Google Scholar] [CrossRef]
- Hou, R.; Zhou, L.; Fu, Y.; Wang, T.; Li, Z.; Zhou, L.; Zhang, G.; Tian, X. Chemical characterization of two fractions from Sanghuangporus sanghuang and evaluation of antidiabetic activity. J. Funct. Foods 2021, 87, 104825. [Google Scholar] [CrossRef]
- Lin, W.; Deng, J.; Huang, S.; Wu, S.; Chen, C.; Lin, W.; Lin, H.; Huang, G. Anti-inflammatory activity of Sanghuangporus sanghuang by suppressing the TLR4-mediated PI3K/AKT/mTOR/IKK beta signaling pathway. RSC Adv. 2017, 18, 21234–21251. [Google Scholar] [CrossRef]
- Keen, N.; Partridg, J.E.; Zaki, A. Pathogen-produced elicitor of a chemical defense mechanism in soybeans monogenically resistant to Phytophthora megasperma var sojae. Phytopathology 1972, 62, 768. [Google Scholar]
- Salehi, M.; Moieni, A.; Safaie, N.; Farhadi, S. Whole fungal elicitors boost paclitaxel biosynthesis induction in Corylus avellana cell culture. PLoS ONE 2020, 15, e0236191. [Google Scholar] [CrossRef]
- Liang, C.; Chen, C.; Zhou, P.; Xu, L.; Zhu, J.; Liang, J.; Zi, J.; Yu, R. Effect of Aspergillus flavus fungal elicitor on the production of terpenoid indole alkaloids in Catharanthus roseus cambial meristematic cells. Molecules 2018, 23, 3276. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, S.; Wang, J.; Li, J.; Liu, D.; Li, J.; Gao, W. Fungal elicitors enhance ginsenosides biosynthesis, expression of functional genes as well as signal molecules accumulation in adventitious roots of Panax ginseng C. A. Mey. J. Biotechnol. 2016, 239, 106–114. [Google Scholar] [CrossRef]
- Prasad, A.; Mathur, A.; Kalra, A.; Gupta, M.M.; Lal, R.K.; Mathur, A.K. Fungal elicitor-mediated enhancement in growth and asiaticoside content of Centella asiatica L. shoot cultures. J. Plant Growth Regul. 2013, 69, 265–273. [Google Scholar] [CrossRef]
- Thomas, S.; Underwood, J.; Tseng, E.; Holloway, A. Long-read sequencing of chicken transcripts and identification of new transcript isoforms. PLoS ONE 2014, 9, e94650. [Google Scholar] [CrossRef]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef]
- Son, S.; Park, Y.; Jung, E.; Singh, D.; Lee, Y.; Kim, J.; Lee, C. Integrated metabolomics and transcriptomics unravel the metabolic pathway variations for different sized beech mushrooms. Int. J. Mol. Sci. 2019, 20, 6007. [Google Scholar] [CrossRef]
- Ku, K.; Becker, T.M.; Juvik, J.A. Transcriptome and metabolome analyses of glucosinolates in two broccoli cultivars following Jasmonate treatment for the induction of glucosinolate defense to Trichoplusia ni (Hübner). Int. J. Mol. Sci. 2016, 17, 1135. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zhang, Q.; Miao, R.; Lin, J.; Feng, R.; Ni, Y.; Li, W.; Yang, D.; Zhao, X. Application of omics technology in the research on edible fungi. Curr. Res. Nutr. Food Sci. 2023, 6, 100430. [Google Scholar] [CrossRef]
- Wang, T.; Wang, G.; Zhang, G.; Hou, R.; Zhou, L.; Tian, X. Systematic analysis of the lysine malonylome in Sanghuangporus sanghuang. BMC Genom. 2021, 22, 840. [Google Scholar] [CrossRef]
- Zhang, G.; Si, J.; Tian, X.; Wang, J. The effects of fungal elicitor on the accumulation of Sanghuangporus sanghuang intracellular metabolites. Mycosystema 2017, 36, 482–491. [Google Scholar] [CrossRef]
- Tao, T.; Li, J. Transformation of FASTA files into feature vectors for unsupervised compression of short reads databases. J. BIionf. Comput. Biol. 2021, 19, 2050048. [Google Scholar] [CrossRef]
- Yang, D.; Liu, Y.; Sun, M.; Zhao, L.; Wang, Y.; Chen, X.; Wei, C.; Gao, L.; Xia, T. Differential gene expression in tea (Camellia sinensis L.) calli with different morphologies and catechin contents. J. Plant Physiol. 2012, 169, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Jeena, G.; Fatima, S.; Tripathi, P.; Upadhyay, S.; Shukla, R. Comparative transcriptome analysis of shoot and root tissue of Bacopa monnieri identifies potential genes related to triterpenoid saponin biosynthesis. BMC Genom. 2017, 18, 490. [Google Scholar] [CrossRef] [PubMed]
- Vitali, C.; Dolcino, M.; Andracco, R.; Pelosi, A.; Fiore, P.; Maglione, W.; Zaccara, E.; Del, P.N.; Puccetti, A. Gene expression profiles in primary Sjögren’s syndrome with and without active systemic manifestations. Clin. Exp. Rheumatol. 2018, 36, 482–491. [Google Scholar] [CrossRef]
- Li, X.; Zhang, J.; Gao, W.; Wang, Y.; Wang, H.; Cao, J.; Huang, L. Chemical composition and anti-inflammatory and antioxidant activities of eight pear cultivars. J. Agr. Food Chem. 2012, 60, 8738–8744. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, B.; Pu, Y.; Tao, J.; Zhang, T. Techniques for the analysis of pentacyclic triterpenoids in medicinal plants. J. Sep. Sci. 2018, 41, 6–19. [Google Scholar] [CrossRef]
- Li, H.; Sun, J.; Xiao, S.; Zhang, L.; Zhou, D. Triterpenoid-Mediated Inhibition of Virus-Host Interaction: Is Now the Time for Discovering Viral Entry/Release Inhibitors from Nature? J. Med. Chem. 2020, 63, 15371–15388. [Google Scholar] [CrossRef]
- Cai, Z.; Kastell, A.; Mewis, I.; Knorr, D.; Smetanska, I. Polysaccharide elicitors enhance anthocyanin and phenolic acid accumulation in cell suspension cultures of Vitis vinifera. Plant Cell Tissue Organ Cult. 2012, 108, 401–409. [Google Scholar] [CrossRef]
- Mercier, L.; Lafitte, C.; Borderies, G.; Briand, X.; Esquerré-Tugayé, M.-T.; Fournier, J. The algal polysaccharide carrageenans can act as an elicitor of plant defense. N. Phytol. 2001, 149, 43–51. [Google Scholar] [CrossRef]
- Cichewicz, R.H.; Kouzi, S.A. Chemistry, biological activity, and chemotherapeutic potential of betulinic acid for the prevention and treatment of cancer and HIV infection. Med. Res. Rev. 2003, 35, 90–114. [Google Scholar] [CrossRef]
- Vaishnav, P.; Demain, A.L. Unexpected applications of secondary metabolites. Biotechnol. Adv. 2011, 29, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Jannet, H.B. Triterpenoids from the fruit barks of Periploca laevigata growing in tunisia. J. Soc. Chim. Tun. 2002, 4, 1565–1569. Available online: https://www.researchgate.net/publication/274632742 (accessed on 1 June 2002).
- Mo, W.; Su, C.; Huang, J.; Liu, J.; Chen, Z.; Cheng, K. Synthesis of acyl oleanolic acid-uracil conjugates and their anti-tumor activity. BMC Chem. 2016, 10, 69. [Google Scholar] [CrossRef]
- Meng, Y.; Zhao, Y.; Kuai, Z.; Liu, L.; Wei, L. Synthesis and antitumor activity evaluation of novel oleanolic acid derivatives. J. Asian Nat. Prod. Res. 2017, 19, 1000–1010. [Google Scholar] [CrossRef]
- Lavanya, S.N.; Udayashankar, A.C.; Raj, S.N.; Mohan, C.D.; Gupta, V.K.; Tarasatyavati, C.; Srivastava, R.; Nayaka, S.C. Lipopolysaccharide-induced priming enhances NO-mediated activation of defense responses in pearl millet challenged with Sclerospora graminicola. 3 Biotech. 2018, 8, 475. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Miao, K.; Zhang, Y.; Pan, S.; Zhang, M.; Jiang, H. Nitric oxide mediates the fungal-elicitor-enhanced biosynthesis of antioxidant polyphenols in submerged cultures of Inonotus obliquus. Microbiology 2009, 155, 3440–3448. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Fu, Y.; Zhang, X.; Wang, T.; Wang, G.; Zhou, L.; Yu, H.; Tian, X. Transcriptome and Metabolome Integration Reveals the Impact of Fungal Elicitors on Triterpene Accumulation in Sanghuangporus sanghuang. J. Fungi 2023, 9, 604. https://doi.org/10.3390/jof9060604
Zhou L, Fu Y, Zhang X, Wang T, Wang G, Zhou L, Yu H, Tian X. Transcriptome and Metabolome Integration Reveals the Impact of Fungal Elicitors on Triterpene Accumulation in Sanghuangporus sanghuang. Journal of Fungi. 2023; 9(6):604. https://doi.org/10.3390/jof9060604
Chicago/Turabian StyleZhou, Linjiang, Yan Fu, Xinyuan Zhang, Tong Wang, Guangyuan Wang, Liwei Zhou, Hailong Yu, and Xuemei Tian. 2023. "Transcriptome and Metabolome Integration Reveals the Impact of Fungal Elicitors on Triterpene Accumulation in Sanghuangporus sanghuang" Journal of Fungi 9, no. 6: 604. https://doi.org/10.3390/jof9060604