
The Chromosome-Scale Genomes of Exserohilum rostratum and Bipolaris zeicola Pathogenic Fungi Causing Rice Spikelet Rot Disease
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Isolates and Genomic DNA Extraction
2.2. Genome Survey and Repeat Sequence Annotation
2.3. Whole-Genome Sequencing and Assembly
2.4. Gene Prediction and Functional Analysis
2.5. Secretome and Effectors Predictionand Toxicity Factor Prediction
2.6. Phylogenetic and Homology Analysis
3. Results
3.1. Pathogen Identification
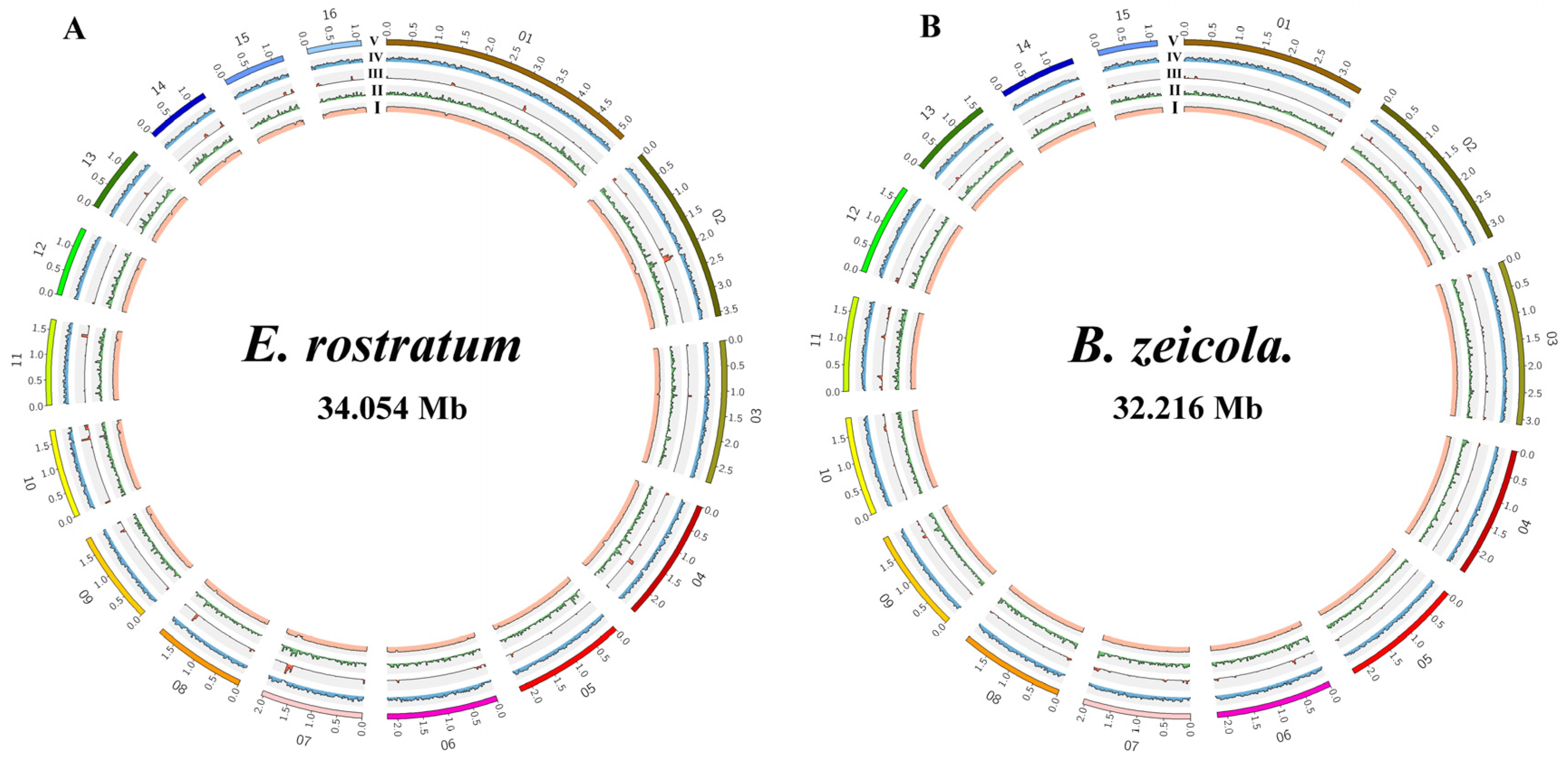
3.2. Genome Sequencing and Assembly
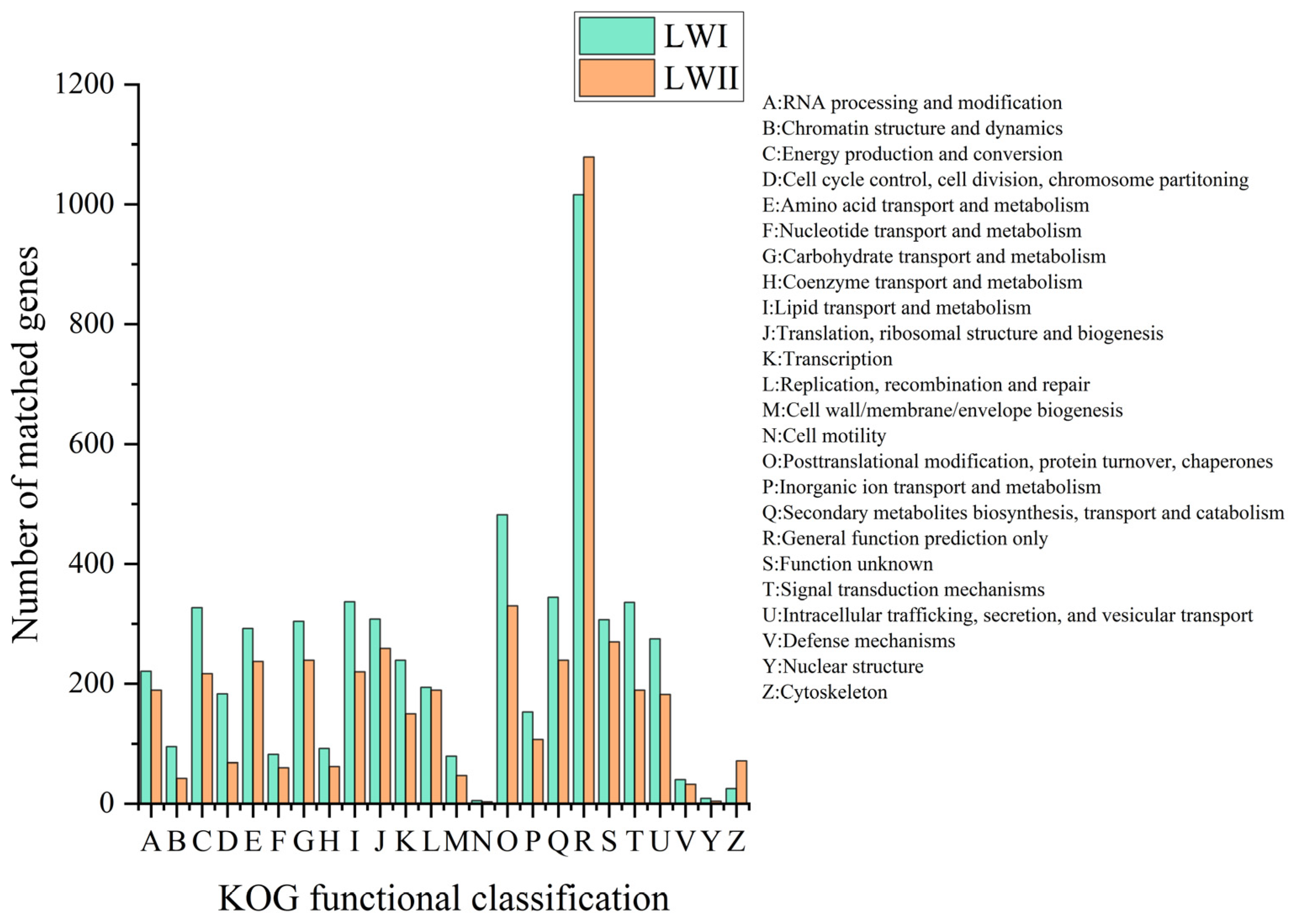
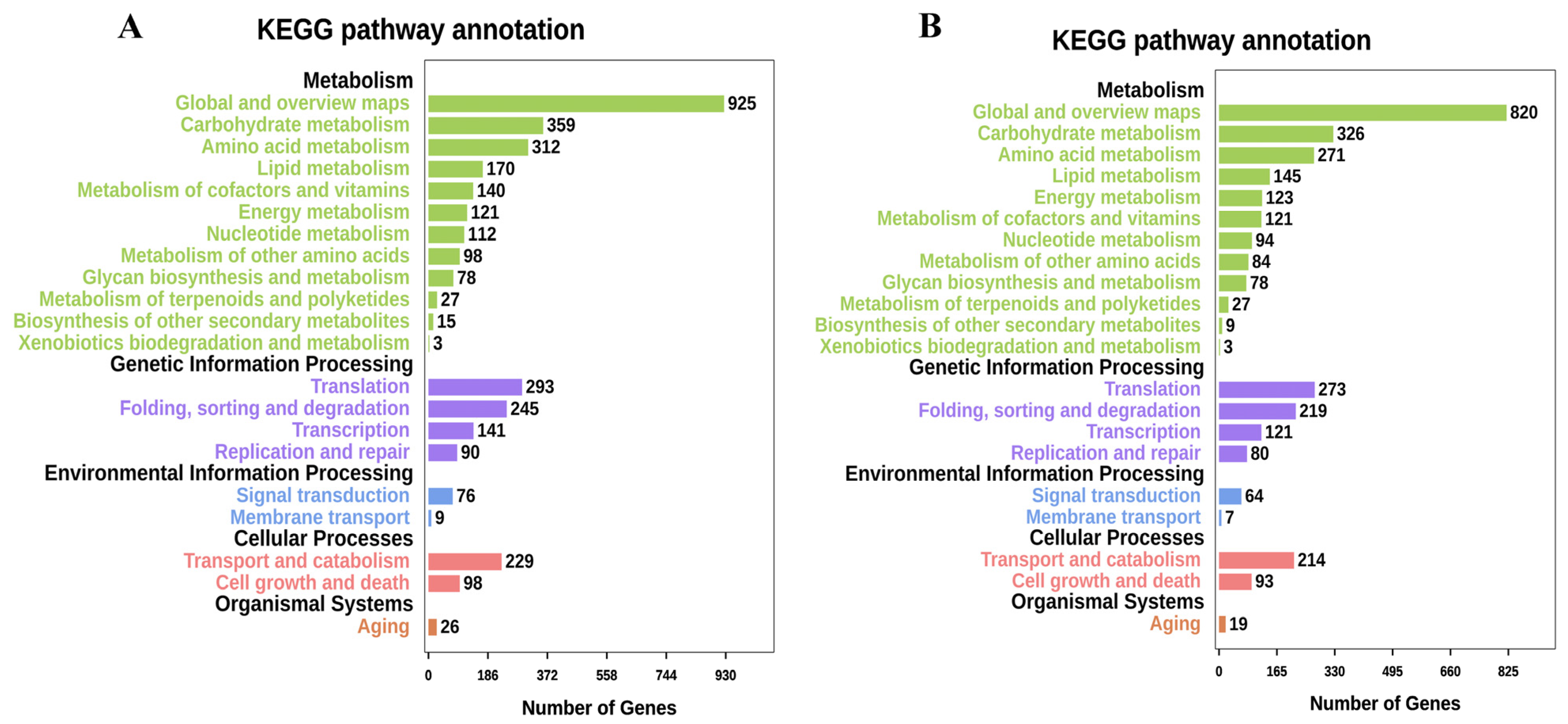
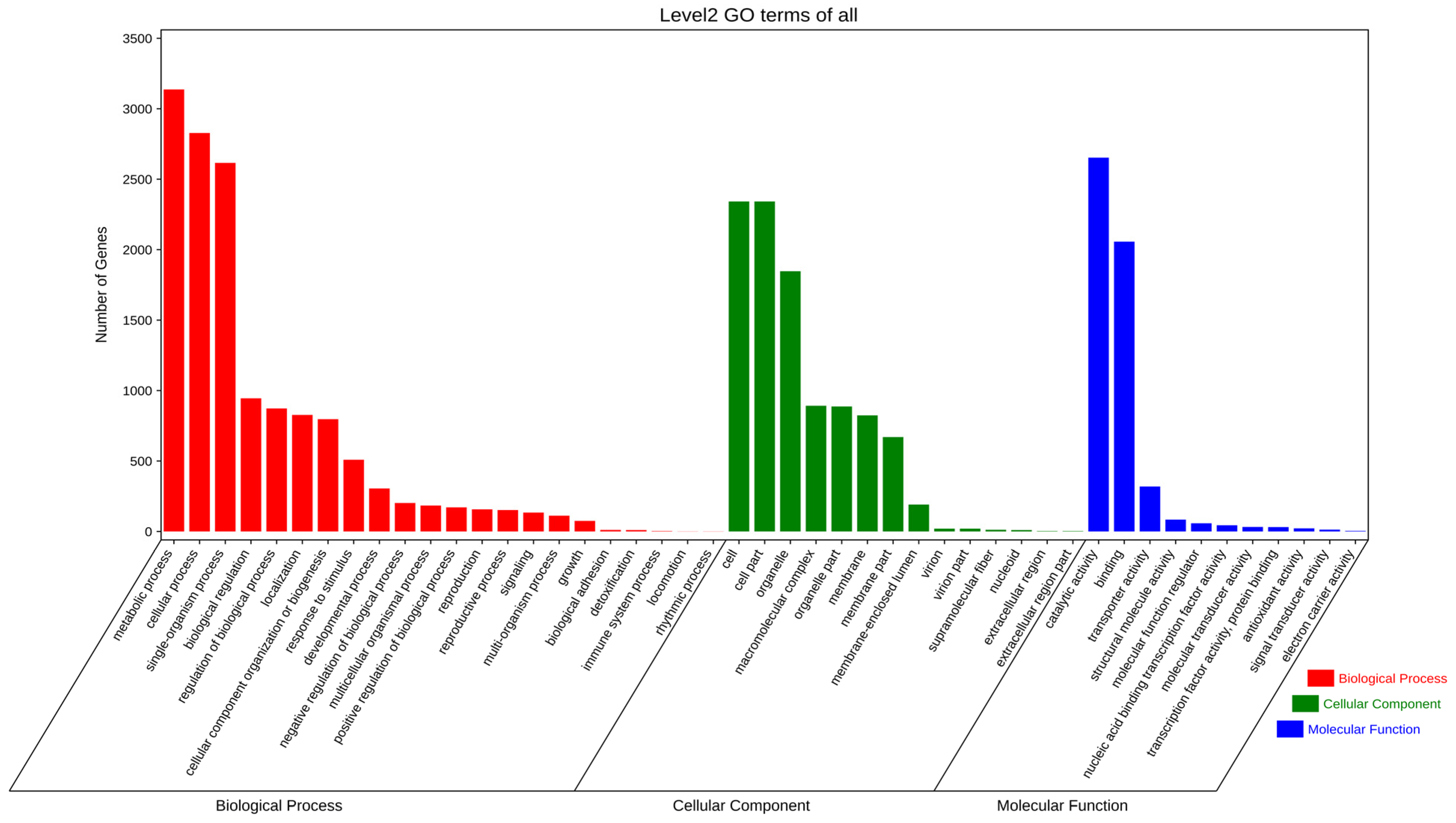
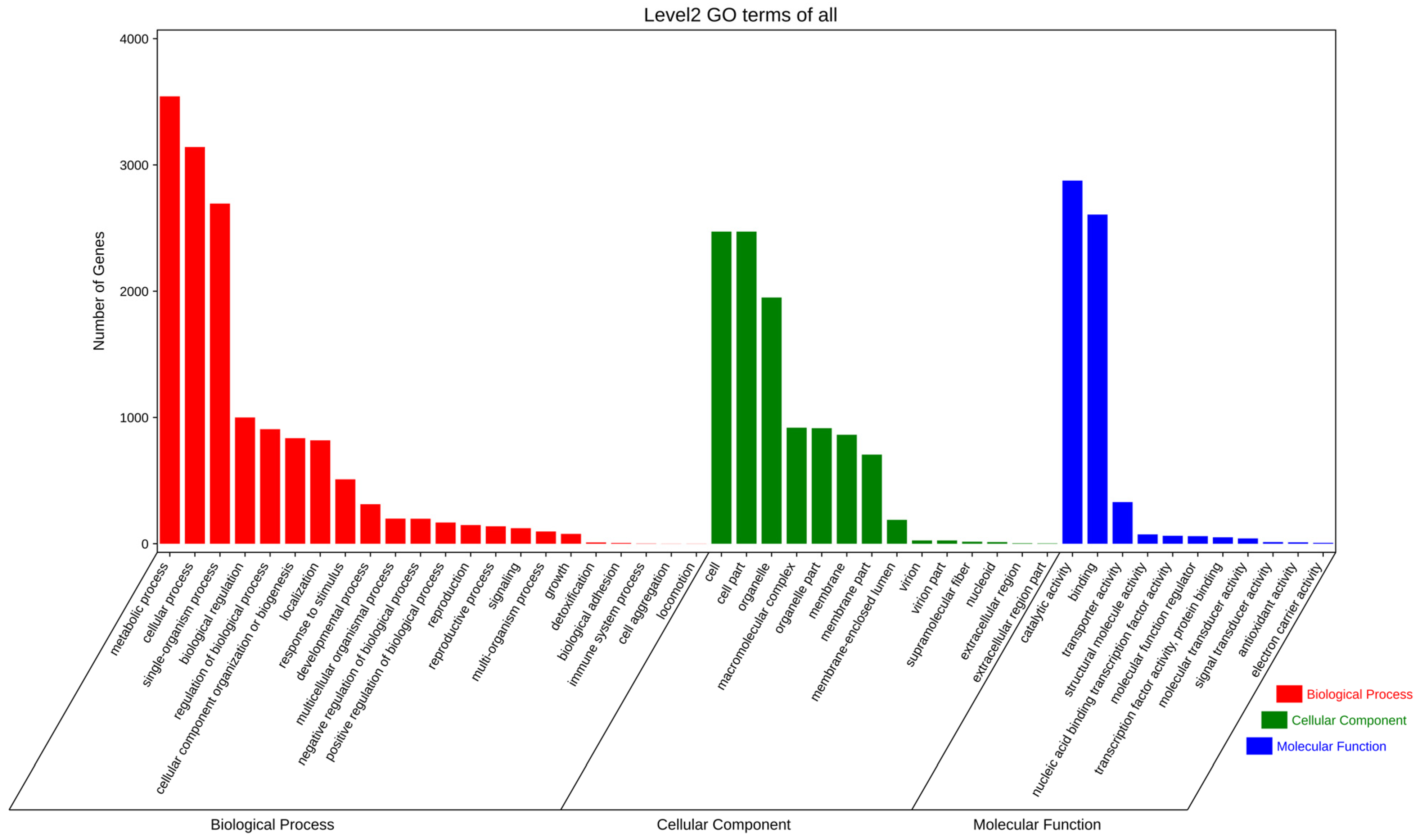
3.3. Gene Prediction and Annotation
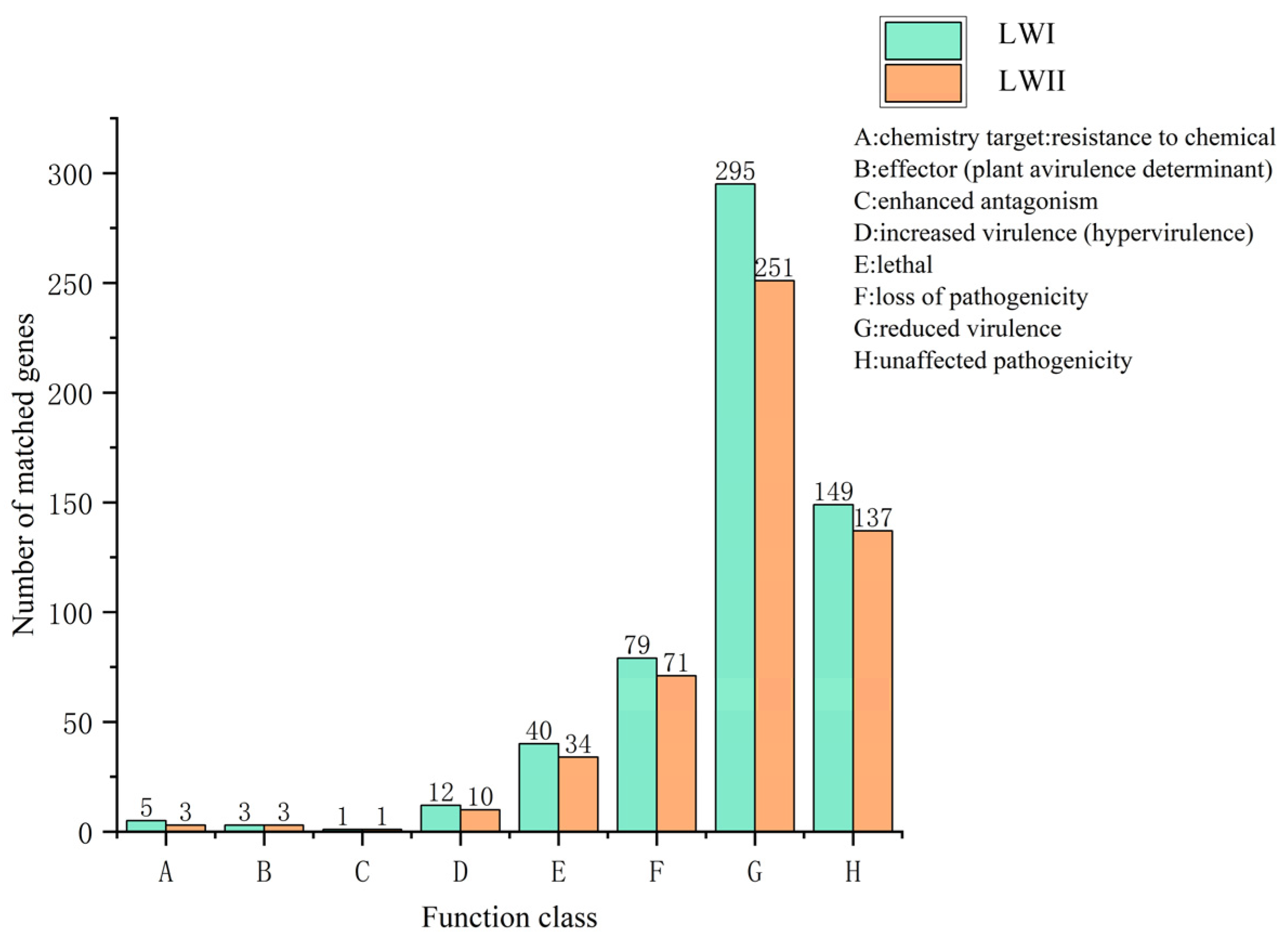
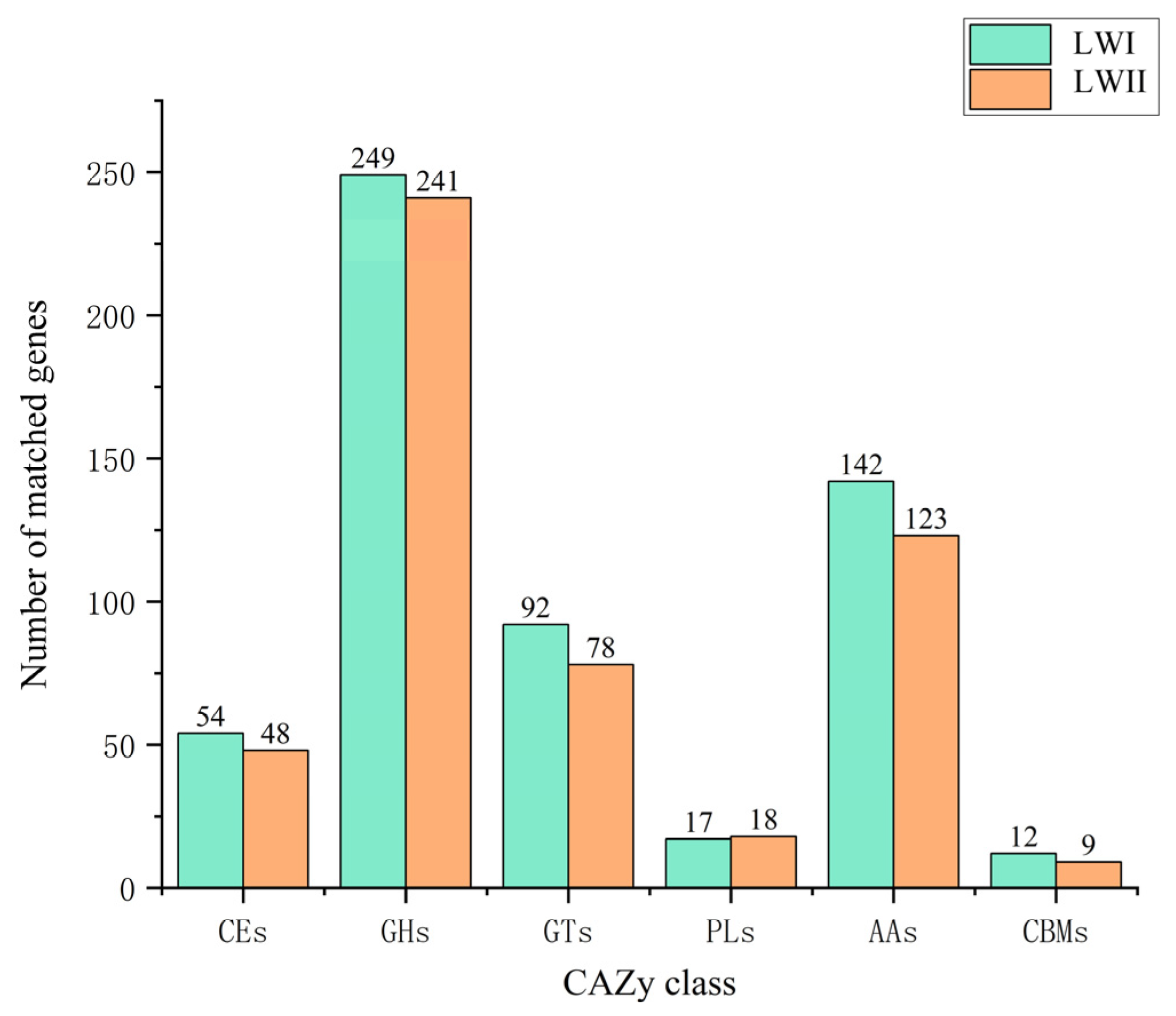
3.4. Prediction and Analysis of Pathogenicity-Related Genes
3.5. Phylogenomics Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, C.; Yang, Y.; Yuan, X.; Xu, Q.; Feng, Y.; Yu, H.; Wang, Y.; Wei, X. Genome-wide association study of blast resistance in India rice. BMC Plant Biol. 2014, 14, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busungu, C.; Taura, S.; Sakagami, J.-I.; Anai, T.; Ichitani, K. High-resolution mapping and characterization of xa42, a resistance gene against multiple Xanthomonas oryzae pv. oryzae races in rice (Oryza sativa L.). Breed. Sci. 2018, 68, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.W.; Wang, L.; Liu, L.M.; Liu, E.Y.; Hou, E.Q.; Xiao, D.F.; Fan, Z.L. Isolation, Identification and Biological Characters of Pathogens of Rice Spikelet Rot Disease. Chin. J. Rice Sci. 2012, 26, 341. [Google Scholar] [CrossRef]
- Zhao, C.Y.; Liu, W.; Guo, Z.J.; Chen, X.J. Identification of pathogens causing brown spot disease on rice glume. Acta Phytophy Sin. 2022, 52, 999–1002. [Google Scholar] [CrossRef]
- Alcorn, J.L. Cochliobolus ellisii sp.nov. Trans. Br. Mycol. Soc. 1983, 81, 172–174. [Google Scholar] [CrossRef]
- Nelson, R.R. Cochliobolus vietoriae, the perfect state of Helminthosporium victoriae. Phytopathology 1960, 50, 774–775. [Google Scholar]
- Hernández-Restrepo, M.; Madrid, H.; Tan, Y.P.; Cunha, K.C.D.; Gené, J.; Guarro, J.; Crous, P.W. Multi-locus phylogeny and taxonomy of Exserohilum. Persoonia 2018, 41, 71–108. [Google Scholar] [CrossRef]
- Gauthier, G.M.; Keller, N.P. Crossover Fungal Pathogens: Crossover fungal pathogens: The biology and pathogenesis of fungi capable of crossing kingdoms to infect plants and humans. Fungal Genet. Biol. 2013, 61, 146–157. [Google Scholar] [CrossRef]
- Graham-Brown, R. Atlas of Clinical Dermatology, 3rd edn. Br. J. Dermatol 2003, 149, 445. [Google Scholar] [CrossRef]
- Yadav, V.; Yang, F.; Reza, M.H.; Liu, S.; Valent, B.; Sanyal, K.; Naqvi, N.I. Cellular Dynamics and Genomic Identity of Centromeres in Cereal Blast Fungus. MBio 2019, 10, e01581-19. [Google Scholar] [CrossRef] [Green Version]
- Manamgoda, D.S.; Cai, L.; McKenzie, E.H.C.; Crous, P.W.; Madrid, H.; Chukeatirote, E.; Shivas, R.G.; Tan, Y.P.; Hyde, K.D. A phylogenetic and taxonomic re-evaluation of the Bipolaris-Cochliobolus-Curvularia Complex. Fungal Divers. 2012, 56, 131–144. [Google Scholar] [CrossRef]
- Praveena, R.; Kozhamburath, A.; Jeevalatha, A.; Bhat, A.I.; Krishnamurthy, K.S. Association of Exserohilum rostratum with ginger: Morphological characterization, phylogenetic relationships and pathogenicity assays. Australas. Plant Path. 2022, 51, 333–344. [Google Scholar] [CrossRef]
- Condon, B.J.; Leng, Y.; Wu, D.; Bushley, K.E.; Ohm, R.A.; Otillar, R.; Martin, J.; Schackwitz, W.; Grimwood, J.; MohdZainudin, N.; et al. Comparative Genome Structure, Secondary Metabolite, and Effector Coding Capacity across Cochliobolus Pathogens. PLoS Genet. 2013, 9, e1003233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X.; Wang, Y.; Zhou, S.; Liu, W.; Wu, H. Genome Sequence Resource for Bipolaris zeicola, the Cause of Northern Corn Leaf Spot Disease. Phytopathology 2022, 112, 1192–1195. [Google Scholar] [CrossRef] [PubMed]
- Litvintseva, A.P.; Hurst, S.; Gade, L.; Frace, M.A.; Hilsabeck, R.; Schupp, J.M.; Gillece, J.D.; Roe, C.; Smith, D.; Keim, P.; et al. Whole-Genome Analysis of Exserohilum rostratum from an Outbreak of Fungal Meningitis and Other Infections. J. Clin. Microbiol. 2014, 52, 3216–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q.; Cheng, C.; Geng, Y.; Zang, R.; Guo, Y.; Yan, L.; Wu, H.; Zhang, M.; Xu, C. The draft genome sequence and characterization of Exserohilum rostratum, a new causal agent of maize leaf spot disease in Chinese Mainland. Eur. J. Plant Pathol. 2023, 165, 57–71. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [Green Version]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B.T. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, R.B.; Liu, B.H.; Xie, Y.L.; Li, Z.Y.; Huang, W.H.; Yuan, J.Y.; He, G.Z.; Chen, Y.X.; Pan, Q.; Liu, Y.J.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 2047–2217X. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, C.L.; Chen, Y.; Xie, S.Q.; Chen, K.N.; Wang, Y.; Luo, F.; Xie, Z. MECAT: An ultra-fast mapping, error correction and de novo assembly tool for single-molecule sequencing reads. Nat. Methods 2017, 14, 1072–1074. [Google Scholar] [CrossRef]
- Hu, J.; Fan, J.P.; Sun, Z.Y.; Liu, S.L. NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Ye, C.X.; Hill, C.M.; Wu, S.G.; Ruan, J.; Ma, Z.S. (Sam). DBG2OLC: Efficient Assembly of Large Genomes Using Long Erroneous Reads of the Third Generation Sequencing Technologies. Sci. Rep. 2016, 6, 31900. [Google Scholar] [CrossRef] [Green Version]
- Delcher, A.L.; Kasif, S.; Fleischmann, R.D.; Peterson, J.; White, O.; Salzberg, S.L. Alignment of whole genomes. Nucleic Acids Res. 1999, 27, 2369–2376. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.Z.; Yang, Z.L.; Busk, P.K.; Xu, Y.; Yin, Y.B. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Yan, C.; Hsu, C.-H.; Chen, Q.-R.; Niu, K.; Komatsoulis, G.A.; Meerzaman, D. OmicCircos: A Simple-to-Use R Package for the Circular Visualization of Multidimensional Omics Data. Cancer Inform. 2014, 13, CIN.S13495. [Google Scholar] [CrossRef] [PubMed]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv, 2022; preprint. [Google Scholar] [CrossRef]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. Advantages of combined transmembrane topology and signal peptide prediction—The Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef] [Green Version]
- Emanuelsson, O.; Nielsen, H.; Brunak, S.; von Heijne, G. Predicting Subcellular Localization of Proteins Based on their N-terminal Amino Acid Sequence. J. Mol. Biol. 2000, 300, 1005–1016. [Google Scholar] [CrossRef] [Green Version]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [Green Version]
- Pierleoni, A.; Martelli, P.L.; Casadio, R. PredGPI: A GPI-anchor predictor. BMC Bioinform. 2008, 9, 392. [Google Scholar] [CrossRef] [Green Version]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; DeSilva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-base: The pathogen–host interactions database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S.O. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A.; Aberer, A.J.; Goll, C.; Smith, S.A.; Berger, S.A.; Izquierdo-Carrasco, F. RAxML-Light: A tool for computing terabyte phylogenies. Bioinformatics 2012, 28, 2064–2066. [Google Scholar] [CrossRef] [Green Version]
- Bashyal, B.M.; Rawat, K.; Sharma, S.; Kulshreshtha, D.; Gopala Krishnan, S.; Singh, A.K.; Dubey, H.; Solanke, A.U.; Sharma, T.R.; Aggarwal, R. Whole Genome Sequencing of Fusarium Fujikuroi Provides Insight into the Role of Secretory Proteins and Cell Wall Degrading Enzymes in Causing Bakanae Disease of Rice. Front. Plant Sci. 2017, 8, 02013. [Google Scholar] [CrossRef] [Green Version]
- Ospina-Giraldo, M.D.; Griffith, J.G.; Laird, E.W.; Mingora, C. The CAZyome of Phytophthora spp.: A comprehensive analysis of the gene complement coding for carbohydrate-active enzymes in species of the genus Phytophthora. BMC Genom. 2010, 11, 525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunings, A.M.; Datnoff, L.E.; Palmateer, A.J.; Locke, J.C.; Krause, C.R. Exserohilum Leaf Spot on Tiger Grass. Plant Health Prog. 2009, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Goss, E.M.; Dickstein, E.R.; Smith, M.E.; Johnson, J.A.; Southwick, F.S.; van Bruggen, A.H.C. Exserohilum rostratum: Characterization of a Cross-Kingdom Pathogen of Plants and Humans. PLoS ONE 2014, 9, e108691. [Google Scholar] [CrossRef]
- Cardona, R.; González, M.S. First Report of Exserohilum rostratum Associated with Rice Seed in Venezuela. Plant Dis. 2007, 91, 226. [Google Scholar] [CrossRef] [Green Version]
- Zibani, A.; Ali, S.; Benslimane, H. Corn diseases in Algeria: First report of three Bipolaris and two Exserohilum species causing leaf spot and leaf blight diseases. Cereal Res. Commun. 2022, 50, 449–461. [Google Scholar] [CrossRef]
- Bennett, H.Y.; Hayes, R.A.; O’Hagan, S. First two cases of Exserohilum rostratum keratitis in Australia. Clin. Exp. Ophthalmol. 2019, 47, 669–671. [Google Scholar] [CrossRef] [PubMed]
- More, S.N.; Hernandez, O.; Castleman, W.L. Mycotic Rhinitis and Sinusitis in Florida Horses. Vet. Pathol. 2019, 56, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Firmino, M.O.; Alves, R.C.; Soares, K.L.; Silva, T.R.; Maruyama, F.H.; Dutra, V.; Galiza, G.J.N.; Dantas, A.F.M. Rhinitis in goat by Exserohilum rostratum (Setosphaeria rostrata). Ciência Rural 2023, 53, e20210807. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Sun, G.Y. Evaluation of intraspecies variation of Exserohilum rostratum by Brn1 gene sequences. Chin. Agric. Sci. Bull. 2005, 21, 66–68. [Google Scholar]
- Kang, I.J.; Shim, H.K.; Roh, J.H.; Heu, S.; Shin, D.B. Simple Detection of Cochliobolus Fungal Pathogens in Maize. Plant Pathol. J. 2018, 34, 327–334. [Google Scholar] [CrossRef]
- Kamchenkov, A.V.; Tsvetkova, Y.V.; Kuznetsova, A.A.; Duchenko, I.P.; Usmanova, G.R. Evaluation of the applicability of classical and molecular methods for diagnosing the phytopathogen Bipolaris zeicola (Stout) Shoemaker in laboratory conditions. Plant Health Quar. 2022, 4, 33–45. [Google Scholar]
- Mapuranga, J.; Chang, J.Y.; Zhang, L.Y.; Zhang, N.; Yang, W.X. Fungal Secondary Metabolites and Small RNAs Enhance Pathogenicity during Plant-Fungal Pathogen Interactions. JoF 2023, 9, 4. [Google Scholar] [CrossRef]
- Brosch, G.; Ransom, R.; Lechner, T.; Walton, J.D.; Loidl, P. Inhibition of maize histone deacetylases by HC toxin, the host-selective toxin of Cochliobolus carbonum. Plant Cell 1995, 7, 1941–1950. [Google Scholar] [CrossRef] [Green Version]
- Ransom, R.F.; Walton, J.D. Histone hyperacetylation in maize in response to treatment with HC-toxin or infection by the filamentous fungus Cochliobolus carbonum. Plant Physiol. 1997, 115, 1021–1027. [Google Scholar] [CrossRef] [Green Version]
- Cipollone, J.; Mourelos, C.; Sisterna, M. First report of Bipolaris zeicola on barley worldwide. Crop Prot. 2020, 135, 105188. [Google Scholar] [CrossRef]
- EI-Shafey, R.A.S.; Attia, K.A.; Mostafa, F.A.; Elamawic, R.M. Incidence and molecular identification of Cochliobolus carbonum as causal organism of rice seedling blight. Beni-Suef Univ. J. Basic Appl. Sci. 2018, 7, 652–662. [Google Scholar] [CrossRef]
- Kang, Z.; Buchenauer, H. Cytology and ultrastructure of the infection of wheat spikes by Fusarium culmorum. Mycol. Res. 2000, 104, 1083–1093. [Google Scholar] [CrossRef]
- Kang, Z.; Buchenauer, H. Ultrastructural and Cytochemical Studies on Cellulose, Xylan and Pectin Degradation in Wheat Spikes Infected by Fusarium culmorum. J. Phytopathol. 2000, 148, 263–275. [Google Scholar] [CrossRef]
- Kang, Z.; Zingen-Sell, I.; Buchenauer, H. Infection of wheat spikes by Fusarium avenaceum and alterations of cell wall components in the infected tissue. Eur. J. Plant Pathol. 2005, 111, 19–28. [Google Scholar] [CrossRef]
- Tyler, L.; Bragg, J.N.; Wu, J.; Yang, X.; Tuskan, G.A.; Vogel, J.P. Annotation and comparative analysis of the glycoside hydrolase genes in Brachypodium distachyon. BMC Genom. 2010, 11, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valent, B.; Khang, C.H. Recent advances in rice blast effector research. Curr. Opin. Plant Biol. 2010, 13, 434–441. [Google Scholar] [CrossRef]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal Effectors and Plant Susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly Feature | LWI | LWII |
|---|---|---|
| Chromosomes | 16 | 15 |
| Total length (bp) | 34,053,972 | 32,215,838 |
| Longest scaffold length (bp) | 5,165,024 | 3,456,296 |
| Contigs N50 (bp) | 2,207,071 | 2,190,445 |
| Contigs N90 (bp) | 1,286,539 | 1,573,589 |
| Genome coverage | 120x | 120x |
| Genome GC % | 50.56 | 50.66 |
| Number of genes | 10,457 | 10,108 |
| Exon average length (bp) | 551 | 583 |
| Exon gene GC% | 34.59 | 33.98 |
| Total gene size (bp) | 5,761,019 | 5,896,621 |
| Database | Annotated Gene Number | Annotation Ratio | ||
|---|---|---|---|---|
| LWI | LWII | LWI | LWII | |
| Nr | 10,245 | 10,079 | 97.97% | 99.71% |
| GO | 4582 | 5282 | 43.82% | 52.26% |
| PHI | 508 | 447 | 0.05% | 0.04% |
| KOG | 5204 | 4485 | 49.77% | 44.37% |
| KEGG | 10,026 | 10,051 | 95.88% | 99.45% |
| Pfam | 7881 | 7966 | 75.42% | 78.81% |
| CAZy | 566 | 517 | 0.05% | 0.05% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, K.; Zhao, C.; Zhang, M.; Li, J.; Zhang, Q.; Wu, X.; Wei, S.; Wang, Y.; Chen, X.; Li, C. The Chromosome-Scale Genomes of Exserohilum rostratum and Bipolaris zeicola Pathogenic Fungi Causing Rice Spikelet Rot Disease. J. Fungi 2023, 9, 177. https://doi.org/10.3390/jof9020177
He K, Zhao C, Zhang M, Li J, Zhang Q, Wu X, Wei S, Wang Y, Chen X, Li C. The Chromosome-Scale Genomes of Exserohilum rostratum and Bipolaris zeicola Pathogenic Fungi Causing Rice Spikelet Rot Disease. Journal of Fungi. 2023; 9(2):177. https://doi.org/10.3390/jof9020177
Chicago/Turabian StyleHe, Ke, Chenyu Zhao, Manman Zhang, Jinshao Li, Qian Zhang, Xiaoyi Wu, Shan Wei, Yong Wang, Xujun Chen, and Cheng Li. 2023. "The Chromosome-Scale Genomes of Exserohilum rostratum and Bipolaris zeicola Pathogenic Fungi Causing Rice Spikelet Rot Disease" Journal of Fungi 9, no. 2: 177. https://doi.org/10.3390/jof9020177