
Pan-Genomics Reveals a New Variation Pattern of Secreted Proteins in Pyricularia oryzae
 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Constructing the Pan-Genome of P. oryzae
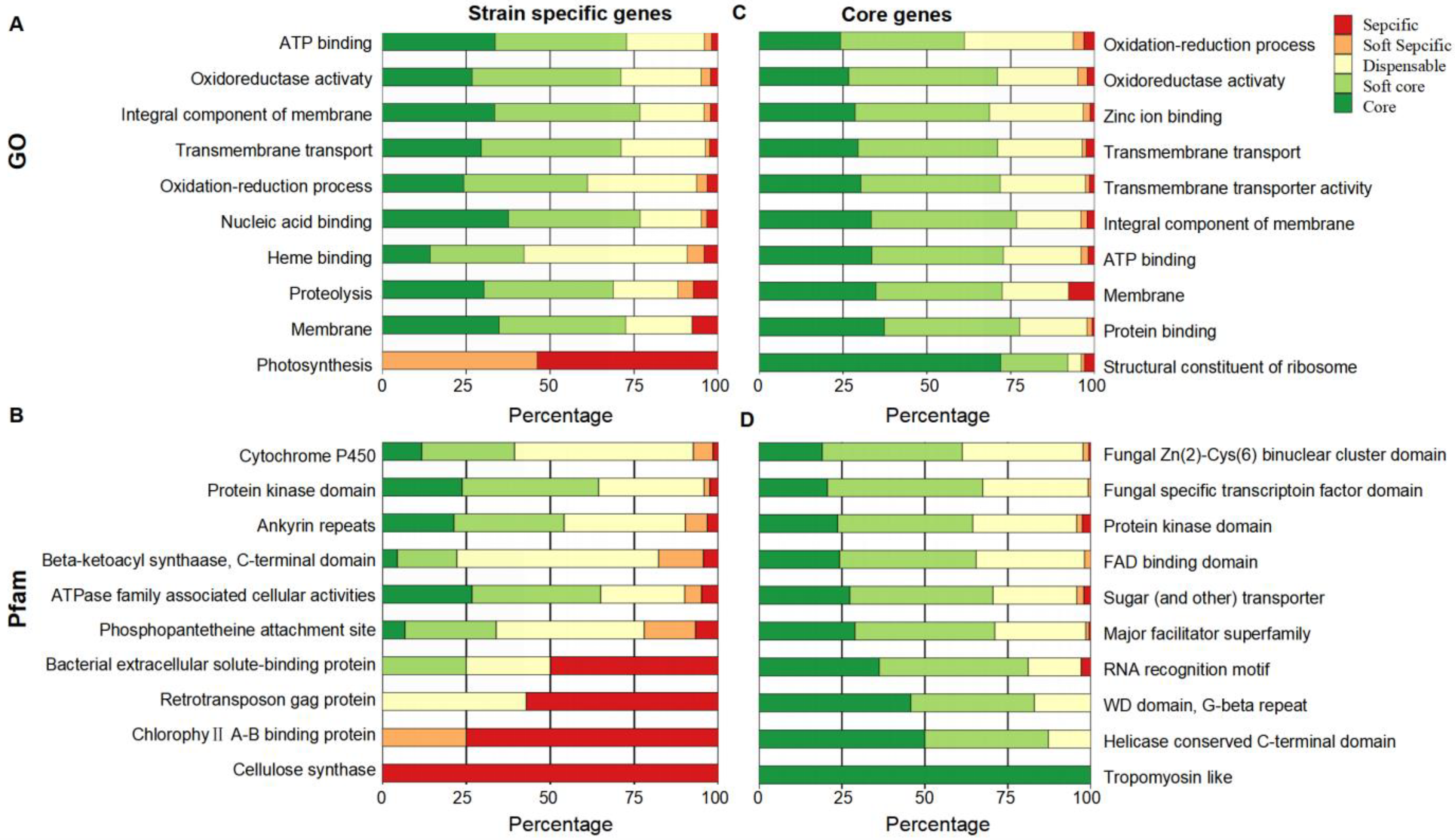
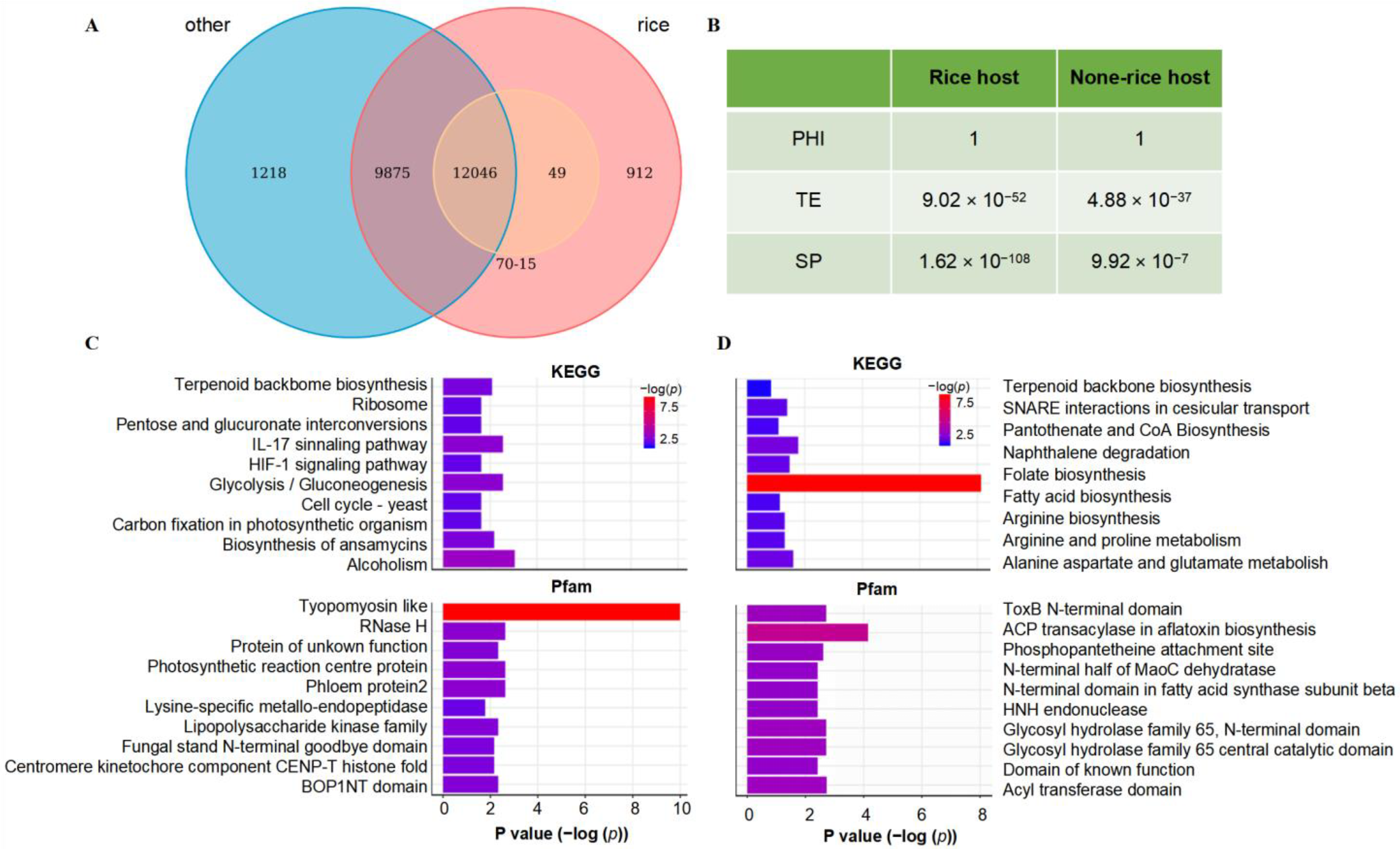
2.2. Functional Enrichment Analysis of Pan-Genes
2.3. Functional Enrichment Analysis of Newly Identified Genes in Pan-Genome
2.4. Functional Analysis of Population Differentiation Related Genes in the Pan-Genome
2.5. Pan-Genomics Reveals a New Variation Pattern of Secreted Proteins in P. oryzae
3. Discussion
4. Materials and Methods
4.1. Genomics Data Collection
4.2. Gene Prediction
4.3. Pan-Genome Construction
4.4. Presence/Absence Variations (PAVs) of Pan-Genes
4.5. Clustering Analysis of Population Structure
4.6. Gene Functional Annotation and Enrichment Analysis
4.7. Population Differentiation Related Genes Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heath, M.C. A generalized concept of host-parasite specificity. Phytopathology 1981, 71, 1121–1123. [Google Scholar] [CrossRef]
- Thierry, M.; Charriat, F.; Milazzo, J.; Adreit, H.; Ravel, S.; Cros-Arteil, S.; Borron, S.; Sella, V.; Kroj, T.; Ioos, R.; et al. Maintenance of divergent lineages of the Rice Blast Fungus Pyricularia oryzae through niche separation, loss of sex and post-mating genetic incompatibilities. PLoS Pathog. 2022, 18, e1010687. [Google Scholar] [CrossRef]
- Inoue, Y.; Vy, T.T.P.; Yoshida, K.; Asano, H.; Mitsuoka, C.; Asuke, S.; Anh, V.L.; Cumagun, C.J.R.; Chuma, I.; Terauchi, R.; et al. Evolution of the wheat blast fungus through functional losses in a host specificity determinant. Science 2017, 357, 80–83. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Mishra, B.; Runge, F.; Thines, M. Gene loss rather than gene gain is associated with a host jump from monocots to dicots in the Smut Fungus Melanopsichium pennsylvanicum. Genome Biol. Evol. 2014, 6, 2034–2049. [Google Scholar] [CrossRef] [Green Version]
- McDonald, B.A.; Linde, C. Pathogen population genetics, evolutionary potential, and durable resistance. Annu. Rev. Phytopathol. 2002, 40, 349–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.J.; van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef] [Green Version]
- Gladieux, P.; Feurtey, A.; Hood, M.E.; Snirc, A.; Clavel, J.; Dutech, C.; Roy, M.; Giraud, T. The population biology of fungal invasions. Mol. Ecol. 2015, 24, 1969–1986. [Google Scholar] [CrossRef]
- de Vienne, D.M.; Giraud, T.; Gouyon, P.H. Lineage Selection and the Maintenance of Sex. PLoS ONE 2013, 8, e66906. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, C.G.P.; Fitzpatrick, D.A. Pan-genome analyses of model fungal species. Microb. Genom. 2019, 5, e000243. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed]
- Sherman, R.M.; Salzberg, S.L. Pan-genomics in the human genome era. Nat. Rev. Genet. 2020, 21, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, B.; Dillon, M.M.; Martel, A.; Almeida, R.N.D.; Desveaux, D.; Guttman, D.S. The pan-genome effector-triggered immunity landscape of a host-pathogen interaction. Science 2020, 367, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Lu, H.; Du, H.; Wang, H.; Chen, W.; Chen, Z.; He, Q.; Ou, S.; Zhang, H.; Li, X.; et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 2021, 184, 3542–3558.e3516. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Shang, L.; Li, X.; He, H.; Yuan, Q.; Song, Y.; Wei, Z.; Lin, H.; Hu, M.; Zhao, F.; Zhang, C.; et al. A super pan-genomic landscape of rice. Cell Res. 2022, 32, 878–896. [Google Scholar] [CrossRef]
- Liu, Y.; Tian, Z. Super graph-based pan-genome: Bringing rice functional genomic study into a new dawn. Mol. Plant 2022, 15, 1409–1411. [Google Scholar] [CrossRef]
- Zhao, Q.; Feng, Q.; Lu, H.; Li, Y.; Wang, A.; Tian, Q.; Zhan, Q.; Lu, Y.; Zhang, L.; Huang, T.; et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018, 50, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L.; et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef]
- Barber, A.E.; Sae-Ong, T.; Kang, K.; Seelbinder, B.; Li, J.; Walther, G.; Panagiotou, G.; Kurzai, O. Aspergillus fumigatus pan-genome analysis identifies genetic variants associated with human infection. Nat. Microbiol. 2021, 6, 1526–1536. [Google Scholar] [CrossRef]
- Horta, M.A.C.; Steenwyk, J.L.; Mead, M.E.; Dos Santos, L.H.B.; Zhao, S.; Gibbons, J.G.; Marcet-Houben, M.; Gabaldon, T.; Rokas, A.; Goldman, G.H. Examination of Genome-Wide Ortholog Variation in Clinical and Environmental Isolates of the Fungal Pathogen Aspergillus fumigatus. mBio 2022, 13, e0151922. [Google Scholar] [CrossRef]
- Kato, R.B.; Jaiswal, A.K.; Tiwari, S.; Barh, D.; Azevedo, V.; Góes-Neto, A. Chapter 12—Pan-genomics of fungi and its applications. In Pan-genomics: Applications, Challenges, and Future Prospects; Barh, D., Soares, S., Tiwari, S., Azevedo, V., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 251–260. [Google Scholar]
- de Jesus Sousa, T.; Jaiswal, A.K.; Hurtado, R.E.; de Oliveira Tosta, S.F.; de Castro Soares, S.; Gomide, A.C.P.; Alcantara, L.C.J.; Barh, D.; Azevedo, V.; Tiwari, S. Chapter 5—Pan-genomics of veterinary pathogens and its applications. In Pan-Genomics: Applications, Challenges, and Future Prospects; Barh, D., Soares, S., Tiwari, S., Azevedo, V., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 101–119. [Google Scholar]
- Valent, B. The Impact of Blast Disease: Past, Present, and Future. Methods Mol. Biol. 2021, 2356, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Nakayashiki, H.; Kataoka, T.; Tamba, H.; Hashimoto, Y.; Tosa, Y.; Mayama, S. Repeat-induced point mutation (RIP) in Magnaporthe grisea: Implications for its sexual cycle in the natural field context. Mol. Microbiol. 2002, 45, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Norvienyeku, J.; Chen, M.; Bao, J.; Lin, L.; Chen, L.; Lin, Y.; Wu, X.; Cai, Z.; Zhang, Q.; et al. Directional Selection from Host Plants Is a Major Force Driving Host Specificity in Magnaporthe Species. Sci. Rep. 2016, 6, 25591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Si, W.; Deng, Q.; Li, P.; Yang, S. Rapid evolution of avirulence genes in rice blast fungus Magnaporthe oryzae. BMC Genet. 2014, 15, 45. [Google Scholar] [CrossRef] [Green Version]
- Chuma, I.; Isobe, C.; Hotta, Y.; Ibaragi, K.; Futamata, N.; Kusaba, M.; Yoshida, K.; Terauchi, R.; Fujita, Y.; Nakayashiki, H.; et al. Multiple translocation of the AVR-Pita effector gene among chromosomes of the rice blast fungus Magnaporthe oryzae and related species. PLoS Pathog. 2011, 7, e1002147. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wang, L.; Wu, W.; He, L.; Yang, X.; Pan, Q. Function and evolution of Magnaporthe oryzae avirulence gene AvrPib responding to the rice blast resistance gene Pib. Sci. Rep. 2015, 5, 11642. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Chen, M.; Zhong, Z.; Tang, W.; Lin, L.; Zhang, X.; Jiang, H.; Zhang, D.; Miao, C.; Tang, H.; et al. PacBio Sequencing Reveals Transposable Elements as a Key Contributor to Genomic Plasticity and Virulence Variation in Magnaporthe oryzae. Mol. Plant 2017, 10, 1465–1468. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Raffaele, S.; Kamoun, S. The two-speed genomes of filamentous pathogens: Waltz with plants. Curr. Opin. Genet. Dev. 2015, 35, 57–65. [Google Scholar] [CrossRef]
- Faino, L.; Seidl, M.F.; Shi-Kunne, X.; Pauper, M.; van den Berg, G.C.; Wittenberg, A.H.; Thomma, B.P. Transposons passively and actively contribute to evolution of the two-speed genome of a fungal pathogen. Genome Res. 2016, 26, 1091–1100. [Google Scholar] [CrossRef] [Green Version]
- Rouxel, T.; Grandaubert, J.; Hane, J.K.; Hoede, C.; van de Wouw, A.P.; Couloux, A.; Dominguez, V.; Anthouard, V.; Bally, P.; Bourras, S.; et al. Effector diversification within compartments of the Leptosphaeria maculans genome affected by Repeat-Induced Point mutations. Nat. Commun. 2011, 2, 202. [Google Scholar] [CrossRef]
- Zhong, Z.; Chen, M.; Lin, L.; Han, Y.; Bao, J.; Tang, W.; Lin, L.; Lin, Y.; Somai, R.; Lu, L.; et al. Population genomic analysis of the rice blast fungus reveals specific events associated with expansion of three main clades. ISME J. 2018, 12, 1867–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plissonneau, C.; Hartmann, F.E.; Croll, D. Pangenome analyses of the wheat pathogen Zymoseptoria tritici reveal the structural basis of a highly plastic eukaryotic genome. BMC Biol. 2018, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Langner, T.; Harant, A.; Gomez-Luciano, L.B.; Shrestha, R.K.; Malmgren, A.; Latorre, S.M.; Burbano, H.A.; Win, J.; Kamoun, S. Genomic rearrangements generate hypervariable mini-chromosomes in host-specific isolates of the blast fungus. PLoS Genet. 2021, 17, e1009386. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Oliveira-Garcia, E.; Lin, G.; Hu, Y.; Dalby, M.; Migeon, P.; Tang, H.; Farman, M.; Cook, D.; White, F.F.; et al. Effector gene reshuffling involves dispensable mini-chromosomes in the wheat blast fungus. PLoS Genet. 2019, 15, e1008272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, M.; Yang, J.; Li, Z.; Hu, S.; Yao, N.; Dean, R.A.; Zhao, W.; Shen, M.; Zhang, H.; Li, C.; et al. Comparative analysis of the genomes of two field isolates of the rice blast fungus Magnaporthe oryzae. PLoS Genet. 2012, 8, e1002869. [Google Scholar] [CrossRef]
- Yoshida, K.; Saitoh, H.; Fujisawa, S.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Tosa, Y.; Chuma, I.; Takano, Y.; Win, J.; et al. Association genetics reveals three novel avirulence genes from the rice blast fungal pathogen Magnaporthe oryzae. Plant Cell 2009, 21, 1573–1591. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Li, Y.; Zhao, M.; Jing, M.; Liu, X.; Liu, M.; Guo, X.; Zhang, X.; Chen, Y.; Liu, Y.; et al. Global genome and transcriptome analyses of Magnaporthe oryzae epidemic isolate 98-06 uncover novel effectors and pathogenicity-related genes, revealing gene gain and lose dynamics in genome evolution. PLoS Pathog. 2015, 11, e1004801. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Huang, H.; Meusnier, I.; Adreit, H.; Ducasse, A.; Bonnot, F.; Pan, L.; He, X.; Kroj, T.; Fournier, E.; et al. Pathogen effectors and plant immunity determine specialization of the blast fungus to rice subspecies. Elife 2016, 5, e19377. [Google Scholar] [CrossRef]
- Hu, Z.J.; Huang, Y.Y.; Lin, X.Y.; Feng, H.; Zhou, S.X.; Xie, Y.; Liu, X.X.; Liu, C.; Zhao, R.M.; Zhao, W.S.; et al. Loss and natural variations of blast fungal avirulence genes breakdown rice resistance genes in the Sichuan Basin of China. Front. Plant Sci. 2022, 13, 788876. [Google Scholar] [CrossRef]
- Fouche, S.; Plissonneau, C.; Croll, D. The birth and death of effectors in rapidly evolving filamentous pathogen genomes. Curr. Opin. Microbiol. 2018, 46, 34–42. [Google Scholar] [CrossRef]
- Zheng, H.; Zhong, Z.; Shi, M.; Zhang, L.; Lin, L.; Hong, Y.; Fang, T.; Zhu, Y.; Guo, J.; Zhang, L.; et al. Comparative genomic analysis revealed rapid differentiation in the pathogenicity-related gene repertoires between Pyricularia oryzae and Pyricularia penniseti isolated from a Pennisetum grass. BMC Genom. 2018, 19, 927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.E.; Kolodner, R.D. A genetic and structural study of genome rearrangements mediated by high copy repeat Ty1 elements. PLoS Genet. 2011, 7, e1002089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvak, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.E.; Thomma, B.; Seidl, M.F. Transposable Elements Contribute to Genome Dynamics and Gene Expression Variation in the Fungal Plant Pathogen Verticillium dahliae. Genome Biol. Evol. 2021, 13, evab135. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Saunders, D.G.; Mitsuoka, C.; Natsume, S.; Kosugi, S.; Saitoh, H.; Inoue, Y.; Chuma, I.; Tosa, Y.; Cano, L.M.; et al. Host specialization of the blast fungus Magnaporthe oryzae is associated with dynamic gain and loss of genes linked to transposable elements. BMC Genom. 2016, 17, 370. [Google Scholar] [CrossRef] [Green Version]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simao, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2021, 2, 100141. [Google Scholar] [CrossRef]
- Pfeifer, B.; Wittelsburger, U.; Ramos-Onsins, S.E.; Lercher, M.J. PopGenome: An efficient Swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 2014, 31, 1929–1936. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Enriched KEGG Pathway | p-Value |
|---|---|---|
| Clade 1 | Lysine biosynthesis | 1.07 × 10−2 |
| Clade 1 | Drug metabolism—other enzymes | 1.07 × 10−2 |
| Clade 1 | Sulfur metabolism | 2.12 × 10−2 |
| Clade 2 | Starch and sucrose metabolism | 7.33 × 10−3 |
| Clade 2 | Glyoxylate and dicarboxylate metabolism | 7.33 × 10−3 |
| Clade 2 | Nitrogen metabolism | 7.33 × 10−3 |
| Clade 3 | Carbon fixation in photosynthetic organisms | 1.94 × 10−3 |
| Clade 3 | Hepatocellular carcinoma | 6.72 × 10−3 |
| Clade 3 | Phenylpropanoid biosynthesis | 1.42 × 10−2 |
| Type | Pfam ID | Pfam Description | p-Value |
|---|---|---|---|
| clade2 vs. clade3 | PF00614 | Phospholipase D Active site motif | 3.68 × 10−4 |
| clade2 vs. clade3 | PF00122 | E1-E2 ATPase | 4.53 × 10−4 |
| clade1 vs. clade2 | PF00443 | Ubiquitin carboxyl-terminal hydrolase | 5.54 × 10−4 |
| clade1 vs. clade2 | PF00082 | Subtilase family | 6.69 × 10−4 |
| clade1 vs. clade3 | PF04082 | Fungal specific transcription factor domain | 1.09 × 10−3 |
| clade1 vs. clade3 | PF00082 | Subtilase family | 1.18 × 10−3 |
| clade2 vs. clade3 | PF00454 | Phosphatidylinositol 3- and 4-kinase | 1.46 × 10−3 |
| clade2 vs. clade3 | PF00702 | haloacid dehalogenase-like hydrolase | 2.16 × 10−3 |
| clade1 vs. clade3 | PF06422 | CDR ABC transporter | 2.33 × 10−3 |
| clade1 vs. clade3 | PF14510 | ABC-transporter N-terminal | 2.33 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, J.; Wang, Z.; Chen, M.; Chen, S.; Chen, X.; Xie, J.; Tang, W.; Zheng, H.; Wang, Z. Pan-Genomics Reveals a New Variation Pattern of Secreted Proteins in Pyricularia oryzae. J. Fungi 2022, 8, 1238. https://doi.org/10.3390/jof8121238
Bao J, Wang Z, Chen M, Chen S, Chen X, Xie J, Tang W, Zheng H, Wang Z. Pan-Genomics Reveals a New Variation Pattern of Secreted Proteins in Pyricularia oryzae. Journal of Fungi. 2022; 8(12):1238. https://doi.org/10.3390/jof8121238
Chicago/Turabian StyleBao, Jiandong, Zhe Wang, Meilian Chen, Shijie Chen, Xiaomin Chen, Jiahui Xie, Wei Tang, Huakun Zheng, and Zonghua Wang. 2022. "Pan-Genomics Reveals a New Variation Pattern of Secreted Proteins in Pyricularia oryzae" Journal of Fungi 8, no. 12: 1238. https://doi.org/10.3390/jof8121238