
Antiproliferative Fatty Acids Isolated from the Polypore Fungus Onnia tomentosa
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Material and Chemicals
2.2. Identification of Mushroom Species
2.3. Preparation of Crude Extracts from Onnia tomentosa
2.4. Fractionation of Extract from Onnia tomentosa
2.5. Cell Line and Assessment for Antiproliferative Activity
2.6. Cell Line and Assessment for Immunomodulatory Activity
2.7. NMR Analyses
2.8. HPLC-MS and HPLC-MS/MS Analyses
2.9. Fatty Acid Methyl Esters Synthesis and GC-FID Quantification
2.10. Autoxidation of Linoleic Acid
3. Results
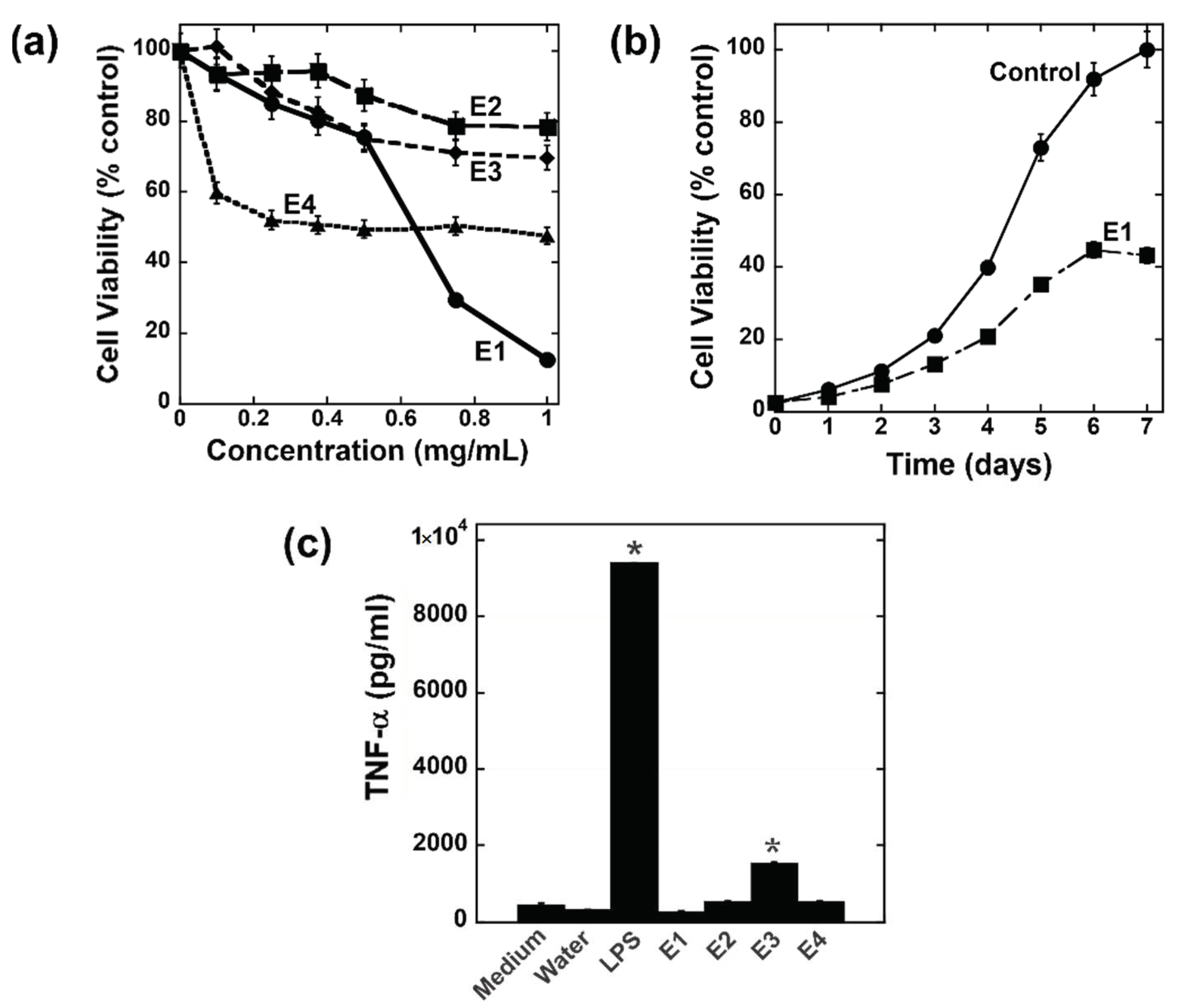
3.1. Assessment of Crude Extracts for Immunomodulatory and Antiproliferative Activities
3.2. Bioactivity-Guided Fractionation and Purification
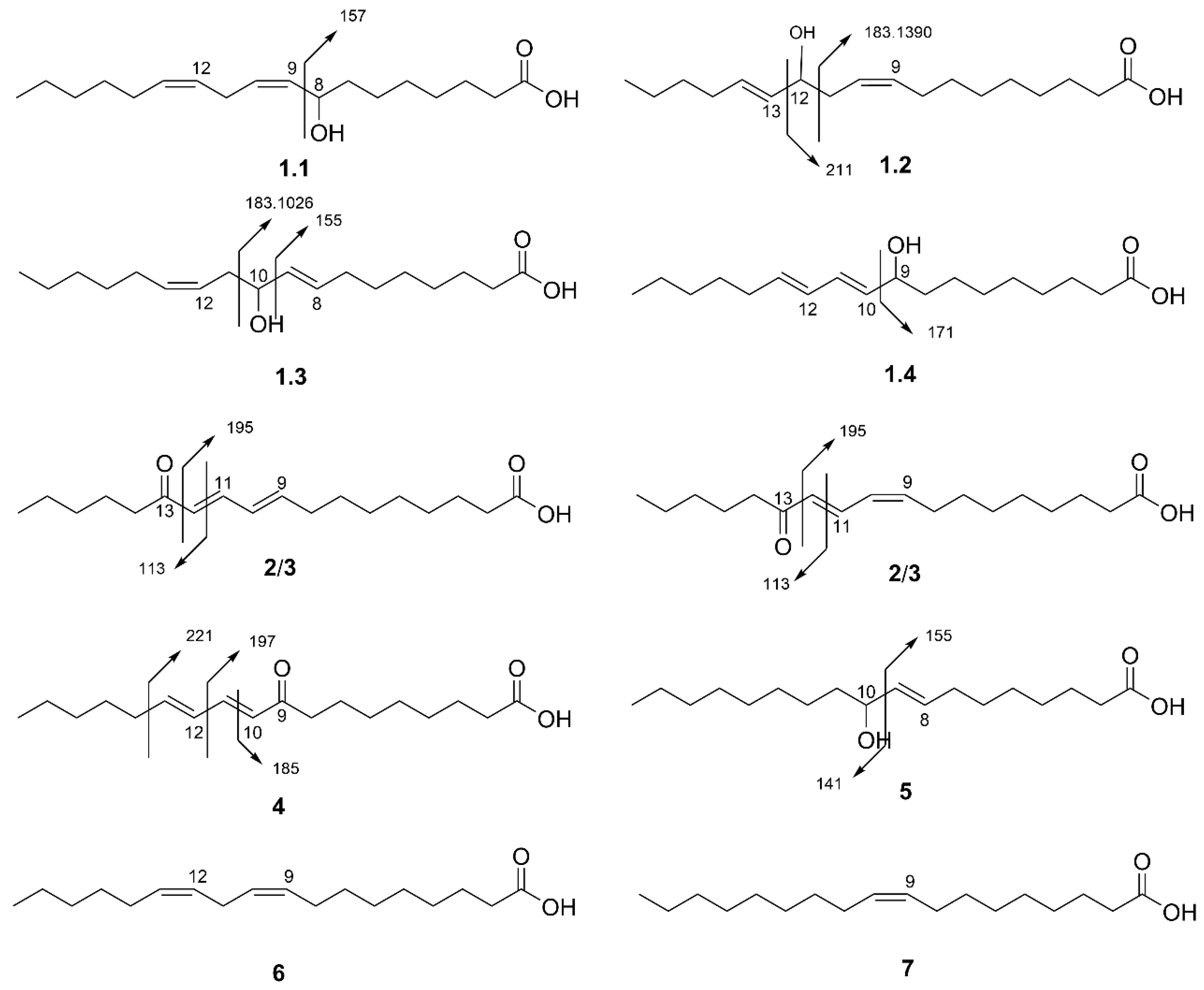
3.3. Identification of Compounds
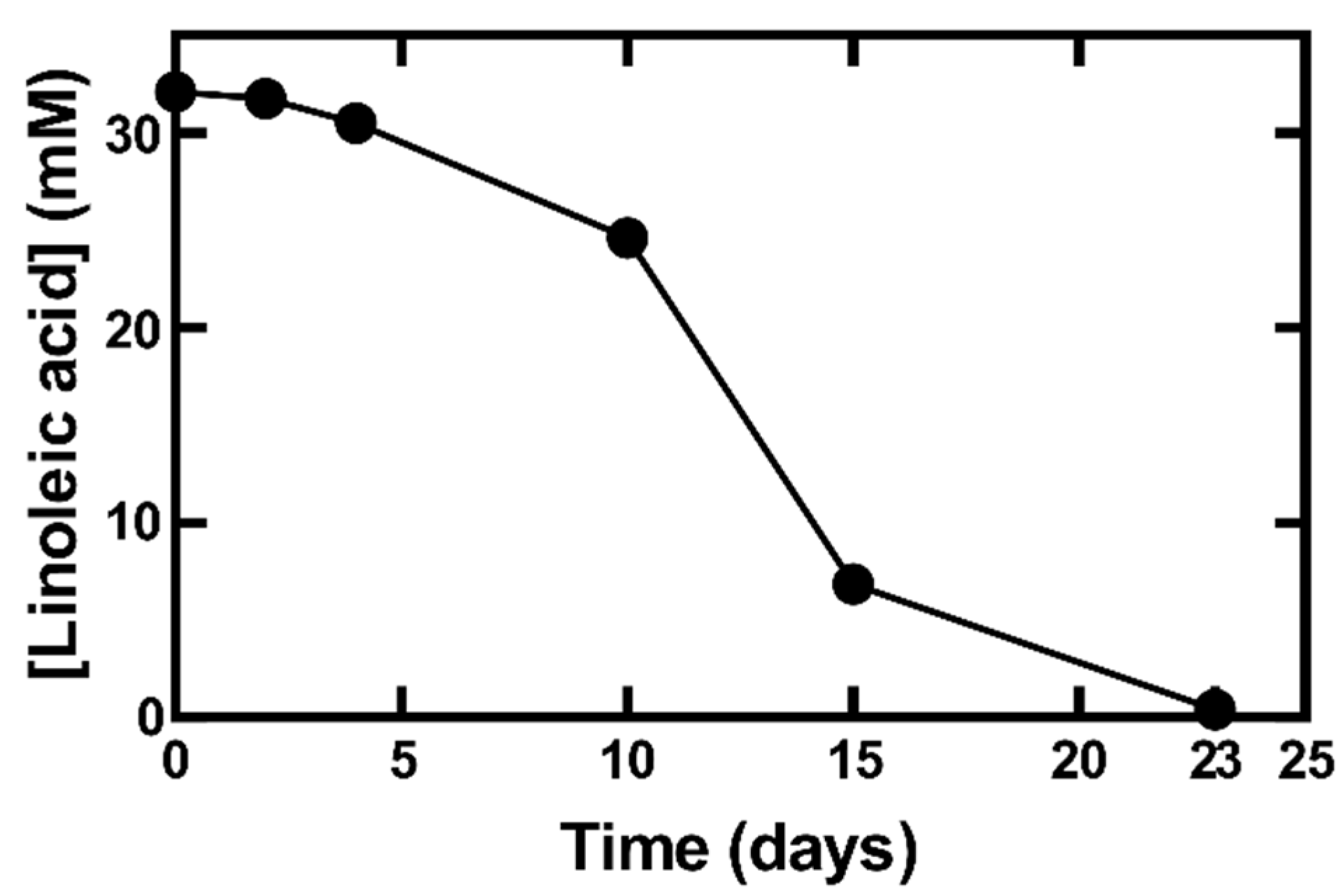
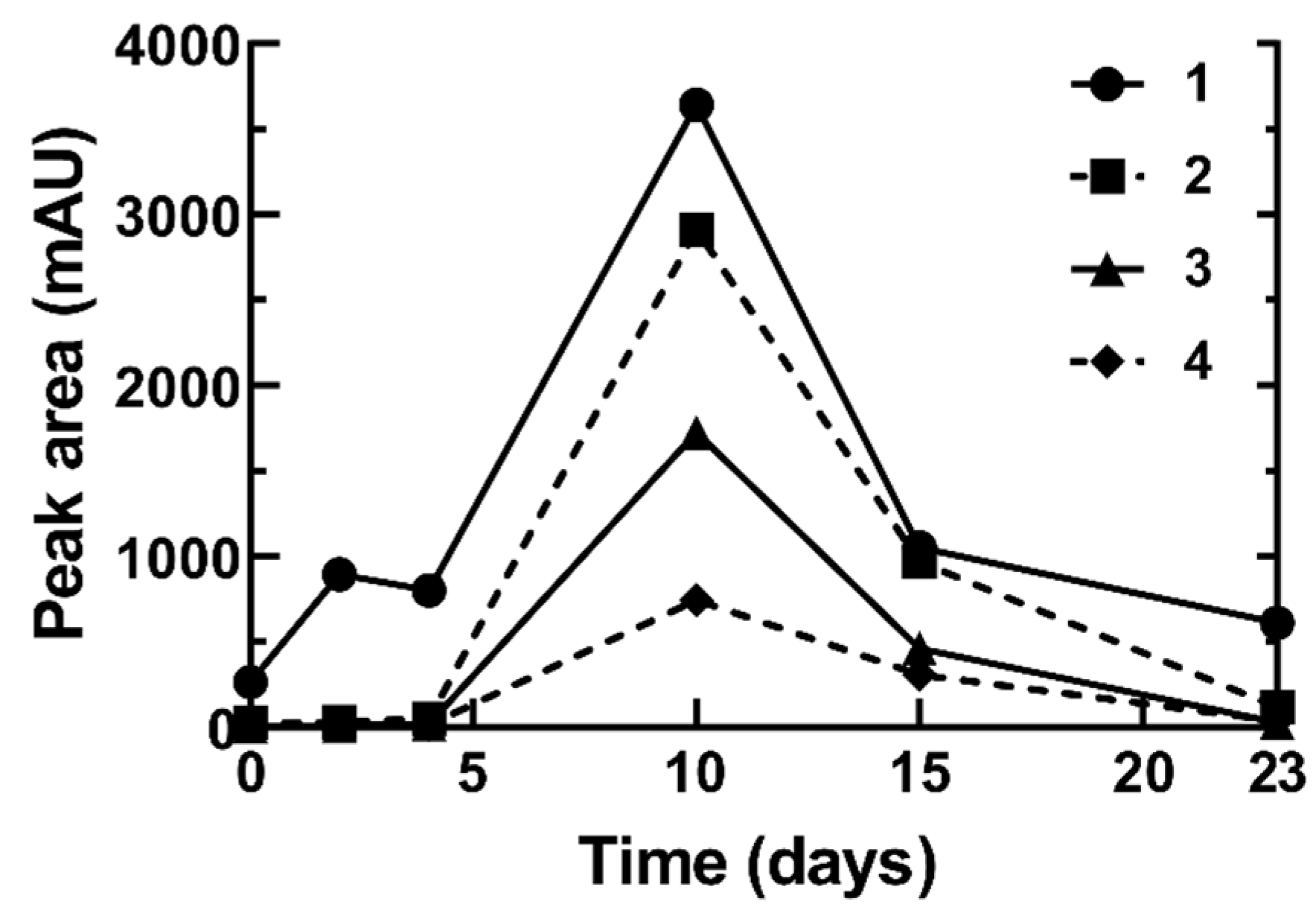
3.4. Autoxidation/Degradation of Linoleic Acid
3.5. Fatty Acid Composition in Selected Mushrooms
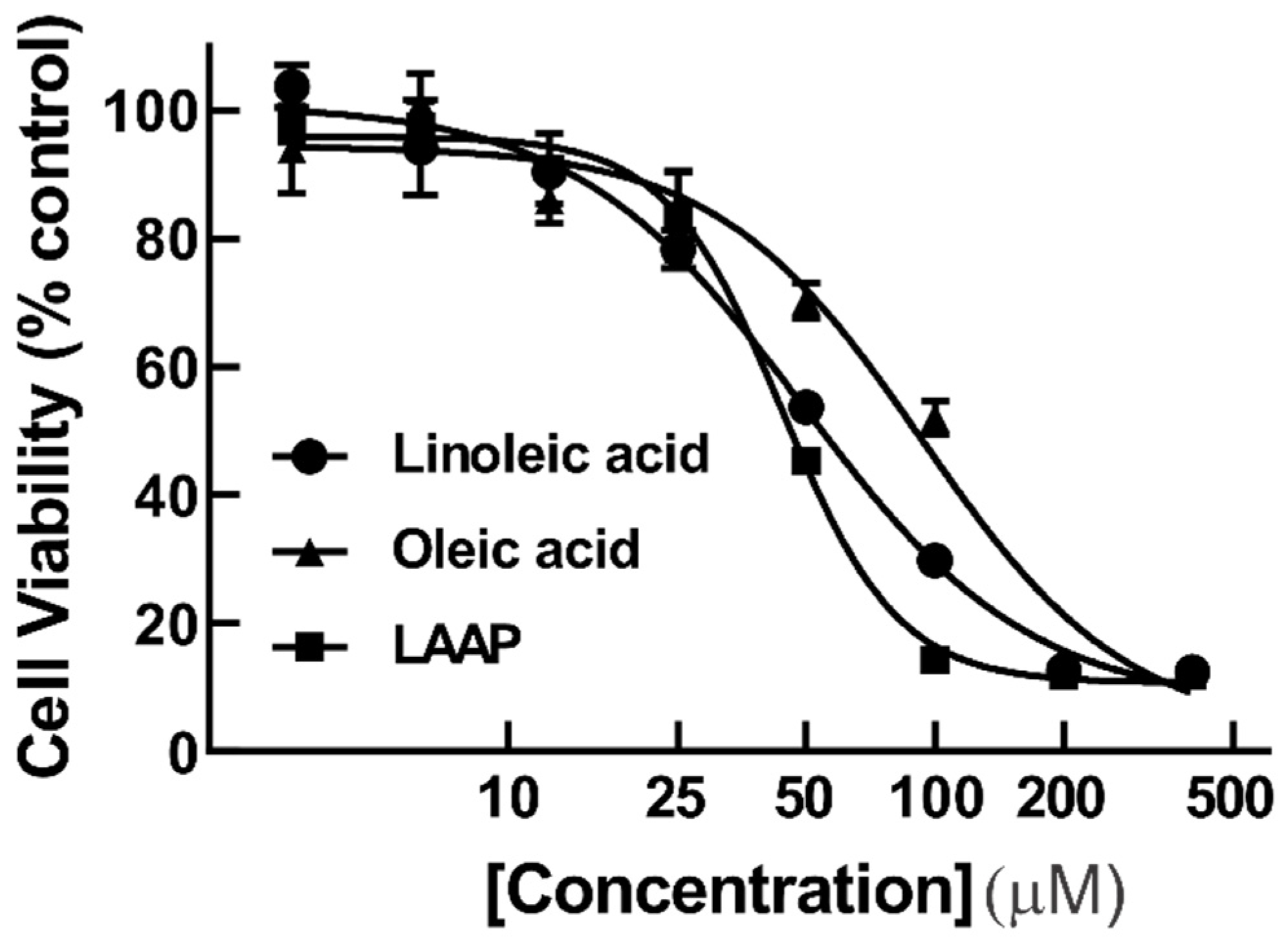
3.6. Antiproliferative Activity of Fatty Acids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zeb, M.; Lee, C.H. Medicinal properties and bioactive compounds from wild mushrooms native to North America. Molecules 2021, 26, 251. [Google Scholar] [CrossRef] [PubMed]
- Barad, A.; Mackedenski, S.; Li, W.M.; Li, X.J.; Lim, B.C.C.; Rashid, F.; Tackaberry, L.E.; Massicotte, H.B.; Egger, K.N.; Reimer, K.; et al. Anti-proliferative activity of a purified polysaccharide isolated from the basidiomycete fungus Paxillus involutus. Carbohydr. Polym. 2018, 181, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Javed, S.; Li, W.M.; Zeb, M.; Yaqoob, A.; Tackaberry, L.E.; Massicotte, H.B.; Egger, K.N.; Cheung, P.C.K.; Payne, G.W.; Lee, C.H. Anti-Inflammatory activity of the wild mushroom, Echinodontium tinctorium, in RAW264.7 macrophage cells and mouse microcirculation. Molecules 2019, 24, 3509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaqoob, A.; Li, W.M.; Liu, V.; Wang, C.; Mackedenski, S.; Tackaberry, L.E.; Massicotte, H.B.; Egger, K.N.; Reimer, K.; Lee, C.H. Grifolin, neogrifolin and confluentin from the terricolous polypore Albatrellus flettii suppress KRAS expression in human colon cancer cells. PLoS ONE 2020, 15, e0231948. [Google Scholar] [CrossRef] [PubMed]
- Zeb, M.; Li, W.M.; Heiss, C.; Black, I.; Tackaberry, L.E.; Massicotte, H.B.; Egger, K.N.; Reimer, K.; Azadi, P.; Lee, C.H. Isolation and characterization of an anti-proliferative polysaccharide from the North American fungus Echinodontium tinctorium. Sci. Rep. 2022, 12, 17298. [Google Scholar] [CrossRef]
- Zeb, M.; Tackaberry, L.E.; Massicotte, H.B.; Egger, K.N.; Reimer, K.; Lu, G.; Heiss, C.; Azadi, P.; Lee, C.H. Structural elucidation and immuno-stimulatory activity of a novel polysaccharide containing glucuronic acid from the fungus Echinodontium tinctorium. Carbohydr. Polym. 2021, 258, 117700. [Google Scholar] [CrossRef]
- Jeitler, M.; Michalsen, A.; Frings, D.; Hübner, M.; Fischer, M.; Koppold-Liebscher, D.A.; Murthy, V.; Kessler, C.S. Significance of medicinal mushrooms in integrative oncology: A narrative review. Front. Pharmacol. 2020, 11, 580656. [Google Scholar] [CrossRef]
- Germain, H.; Bergeron, M.-J.; Bernier, L.; Laflamme, G.; Hamelin, R.C. Patterns of colonization and spread in the fungal spruce pathogen Onnia tomentosa. Mol. Ecol. 2009, 18, 4422–4433. [Google Scholar] [CrossRef]
- Laflamme, G. Root diseases in forest ecosystems. Can. J. Plant Pathol. 2010, 32, 68–76. [Google Scholar] [CrossRef]
- Bernier, D.R. Site and Soil Characteristics Related to the Incidence and Spread of Inonotus tomentosus. Master’s Thesis, University of Northern British Columbia, Prince George, BC, Canada, 1997. [Google Scholar]
- Deo, G.S.; Khatra, J.; Buttar, S.; Li, W.M.; Tackaberry, L.E.; Massicotte, H.B.; Egger, K.N.; Reimer, K.; Lee, C.H. Antiproliferative, immunostimulatory, and anti-inflammatory activities of extracts derived from mushrooms collected in Haida Gwaii, British Columbia (Canada). Int. J. Med. Mushrooms 2019, 21, 629–643. [Google Scholar] [CrossRef]
- Smith, A.; Javed, S.; Barad, A.; Myhre, V.; Li, W.M.; Reimer, K.; Massicotte, H.B.; Tackaberry, L.E.; Payne, G.W.; Egger, K.N.; et al. Growth-inhibitory and immunomodulatory activities of wild mushrooms from North-Central British Columbia (Canada). Int. J. Med. Mushrooms 2017, 19, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.W.; Holroyd, C.M.; Aguilar, J.A.; Nilsson, M.; Morris, G.A. “Perfecting” WATERGATE: Clean proton NMR spectra from aqueous solution. Chem. Commun. 2013, 49, 358–360. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Morrison, W.R.; Smith, L.M. Preparation of fatty acid methyl esters and dimethylacetals from lipids with boron fluoride–methanol. J. Lipid Res. 1964, 5, 600–608. [Google Scholar] [CrossRef]
- Holub, B.J.; Wlodek, M.; Rowe, W.; Piekarski, J. Correlation of omega-3 levels in serum phospholipid from 2053 human blood samples with key fatty acid ratios. Nutr. J. 2009, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Koshino, H.; Togiya, S.; Yoshihara, T.; Sakamura, S.; Shimanuki, T.; Sato, T.; Tajimi, A. Four fungitoxic C-18 hydroxy unsaturated fatty acids from stromata of Epichloe typhina. Tetrahedron Lett. 1987, 28, 73–76. [Google Scholar] [CrossRef]
- Rao, A.V.R.; Reddy, E.R.; Purandare, A.V.; Varaprasad, C.V.N.S. Stereoselective synthesis of unsaturated C-18 hydroxy fatty acids the self defensive substances. Tetrahedron 1987, 43, 4385–4394. [Google Scholar] [CrossRef]
- Yokoi, H.; Mizukami, H.; Nagatsu, A.; Ohno, T.; Tanabe, H.; Inoue, M. Peroxisome proliferator-activated receptor γ Ligands isolated from Adlay seed (Coix lacryma-jobi L. var. ma-yuen STAPF.). Biol. Pharm. Bull. 2009, 32, 735–740. [Google Scholar] [CrossRef] [Green Version]
- Trapp, M.A.; Kai, M.; Mithöfer, A.; Rodrigues-Filho, E. Antibiotic oxylipins from Alternanthera brasiliana and its endophytic bacteria. Phytochemistry 2015, 110, 72–82. [Google Scholar] [CrossRef]
- Garscha, U.; Oliw, E.H. Steric analysis of 8-hydroxy- and 10-hydroxyoctadecadienoic acids and dihydroxyoctadecadienoic acids formed from 8R-hydroperoxyoctadecadienoic acid by hydroperoxide isomerases. Anal. Biochem. 2007, 367, 238–246. [Google Scholar] [CrossRef]
- Oliw, E.H.; Su, C.; Skogström, T.; Benthin, G. Analysis of novel hydroperoxides and other metabolites of oleic, linoleic, and linolenic acids by liquid chromatography-mass spectrometry with ion trap MSn. Lipids 1998, 33, 843–852. [Google Scholar] [CrossRef]
- Yoshida, Y.; Kodai, S.; Takemura, S.; Minamiyama, Y.; Niki, E. Simultaneous measurement of F2-isoprostane, hydroxyoctadecadienoic acid, hydroxyeicosatetraenoic acid, and hydroxycholesterols from physiological samples. Anal. Biochem. 2008, 379, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.; Loonis, M. Regio- and stereoselective oxidation of linoleic acid bound to serum albumin: Identification by ESI–mass spectrometry and NMR of the oxidation products. Chem. Phys. Lipids 2005, 138, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Barros, L.; Baptista, P.; Correia, D.M.; Casal, S.; Oliveira, B.; Ferreira, I.C. Fatty acid and sugar compositions, and nutritional value of five wild edible mushrooms from Northeast Portugal. Food Chem. 2007, 105, 140–145. [Google Scholar] [CrossRef]
- Çayan, F.; Deveci, E.; Tel-Çayan, G.; Duru, M.E. Chemometric approaches for the characterization of the fatty acid composition of seventeen mushroom species. Anal. Lett. 2020, 53, 2784–2798. [Google Scholar] [CrossRef]
- Marekov, I.; Momchilova, S.; Grung, B.; Nikolova-Damyanova, B. Fatty acid composition of wild mushroom species of order Agaricales—Examination by gas chromatography–mass spectrometry and chemometrics. J. Chromatogr. B 2012, 910, 54–60. [Google Scholar] [CrossRef]
- Banni, S.; Contini, M.S.; Angioni, E.; Deiana, M.; Dessì, M.A.; Melis, M.P.; Carta, G.; Corongiu, F.P. A novel approach to study linoleic acid autoxidation: Importance of simultaneous detection of the substrate and its derivative oxidation products. Free Radic. Res. 1996, 25, 43–53. [Google Scholar] [CrossRef]
- Günç Ergönül, P.; Akata, I.; Kalyoncu, F.; Ergönül, B. Fatty acid compositions of six wild edible mushroom species. Sci. World J. 2013, 2013, 163964. [Google Scholar] [CrossRef] [Green Version]
- Akazawa-Ogawa, Y.; Shichiri, M.; Nishio, K.; Yoshida, Y.; Niki, E.; Hagihara, Y. Singlet-oxygen-derived products from linoleate activate Nrf2 signaling in skin cells. Free Radic. Biol. Med. 2015, 79, 164–175. [Google Scholar] [CrossRef]
- Yuan, Z.-X.; Rapoport, S.I.; Soldin, S.J.; Remaley, A.T.; Taha, A.Y.; Kellom, M.; Gu, J.; Sampson, M.; Ramsden, C.E. Identification and profiling of targeted oxidized linoleic acid metabolites in rat plasma by quadrupole time-of-flight mass spectrometry. Biomed. Chromatogr. 2013, 27, 422–432. [Google Scholar] [CrossRef]
- Culleré, J.; Durany, O.; Busquets, M.; Manresa, A. Biotransformation of oleic acid into (E)-10-hydroxy-8-octadecenoic acid and (E)-7,10-dihydroxy-8-octadecenoic acid by Pseudomonas sp. 42A2 in an immobilized system. Biotechnol. Lett. 2001, 23, 215–219. [Google Scholar] [CrossRef]
- De Andres, C.; Mercade, E.; Guinea, J.; Manresa, A. 7, 10-Dihydroxy-8 (E)-octadecenoic acid produced by Pseudomonas 42A2: Evaluation of different cultural parameters of the fermentation. World J. Microbiol. Biotechnol. 1994, 10, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.T.; Bagby, M.O. 10-Hydroxy-8 (Z)-octadecenoic acid, an intermediate in the bioconversion of oleic acid to 7, 10-dihydroxy-8 (E)-octadecenoic acid. J. Ind. Microbiol. 1992, 9, 103–107. [Google Scholar] [CrossRef]
- Kim, H.; Gardner, H.W.; Hou, C.T. 10 (S)-hydroxy-8 (E)-octadecenoic acid, an intermediate in the conversion of oleic acid to 7, 10-dihydroxy-8 (E)-octadecenoic acid. J. Am. Oil Chem. Soc. 2000, 77, 95–99. [Google Scholar] [CrossRef]
- Kuo, T.M.; Nakamura, L.K. Diversity of oleic acid, ricinoleic acid and linoleic acid conversions among Pseudomonas aeruginosa strains. Curr. Microbiol. 2004, 49, 261–266. [Google Scholar] [CrossRef]
- Wadman, M.W.; van Zadelhoff, G.; Hamberg, M.; Visser, T.; Veldink, G.A.; Vliegenthart, J.F. Conversion of linoleic acid into novel oxylipins by the mushroom Agaricus bisporus. Lipids 2005, 40, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.R.; Akpinar, A.; Prabhune, A.A.; Friend, J.; Ratledge, C. The biosynthesis of oxylipins of linoleic and arachidonic acids by the sewage fungus Leptomitus lacteus, including the identification of 8R-hydroxy-9Z, 12Z-octadecadienoic acid. Lipids 2000, 35, 23–30. [Google Scholar] [CrossRef]
- Sagar, P.S.; Das, U.N. Cytotoxic action of cis-unsaturated fatty acids on human cervical carcinoma (HeLa) cells in vitro. Prostaglandins Leukot Essent Fatty Acids 1995, 53, 287–299. [Google Scholar] [CrossRef]
- Dommels, Y.E.; Haring, M.M.; Keestra, N.G.; Alink, G.M.; van Bladeren, P.J.; van Ommen, B. The role of cyclooxygenase in n-6 and n-3 polyunsaturated fatty acid mediated effects on cell proliferation, PGE(2) synthesis and cytotoxicity in human colorectal carcinoma cell lines. Carcinogenesis 2003, 24, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.; Shen, Y.; Shen, J.; Zhou, F.; Shen, S.; Das, U.N. Effect of n-3 and n-6 unsaturated fatty acids on prostate cancer (PC-3) and prostate epithelial (RWPE-1) cells in vitro. Lipids Health Dis. 2013, 12, 160. [Google Scholar] [CrossRef]
- Mouradian, M.; Kikawa, K.D.; Johnson, E.D.; Beck, K.L.; Pardini, R.S. Key roles for GRB2-associated-binding protein 1, phosphatidylinositol-3-kinase, cyclooxygenase 2, prostaglandin E2 and transforming growth factor alpha in linoleic acid-induced upregulation of lung and breast cancer cell growth. Prostaglandins Leukot Essent Fatty Acids 2014, 90, 105–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Su, C.; Luo, X.; Zeng, H.; Zhao, L.; Wei, L.; Zhang, X.; Varghese, Z.; Moorhead, J.F.; Chen, Y. Dietary oleic acid-induced CD36 promotes cervical cancer cell growth and metastasis via up-regulation Src/ERK pathway. Cancer Lett. 2018, 438, 76–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dailey, O.D.; Wang, X.; Chen, F.; Huang, G. Anticancer activity of branched-chain derivatives of oleic acid. Anticancer Res. 2011, 31, 3165–3169. [Google Scholar] [PubMed]
- Liotti, A.; Cosimato, V.; Mirra, P.; Calì, G.; Conza, D.; Secondo, A.; Luongo, G.; Terracciano, D.; Formisano, P.; Beguinot, F. Oleic acid promotes prostate cancer malignant phenotype via the G protein-coupled receptor FFA1/GPR40. J. Cell. Physiol. 2018, 233, 7367–7378. [Google Scholar] [CrossRef]
- Storniolo, C.E.; Martínez-Hovelman, N.; Martínez-Huélamo, M.; Lamuela-Raventos, R.M.; Moreno, J.J. Extra virgin olive oil minor compounds modulate mitogenic action of oleic acid on colon cancer cell line. J. Agric. Food Chem. 2019, 67, 11420–11427. [Google Scholar] [CrossRef] [PubMed]
- Sande, D.; de Oliveira, G.P.; e Moura, M.A.F.; de Almeida Martins, B.; Lima, M.T.N.S.; Takahashi, J.A. Edible mushrooms as a ubiquitous source of essential fatty acids. Food Res. Int. 2019, 125, 108524. [Google Scholar] [CrossRef] [PubMed]
- Germain, H.; Laflamme, G.; Bernier, L.; Boulet, B.; Hamelin, R.C. DNA polymorphism and molecular diagnosis in Inonotus spp. Can. J. Plant Pathol. 2002, 24, 194–199. [Google Scholar] [CrossRef]
- Gottlieb, A.M.; Wright, J.E.; Moncalvo, J.-M. Inonotus sl in Argentina-morphology, cultural characters and molecular analyses. Mycol. Prog. 2002, 1, 299–313. [Google Scholar] [CrossRef]
- Jin, C.-Y.; Jeong, M.-J.; Kim, G.-Y.; Park, J.-M.; Kim, M.-O.; Moon, D.-O.; Lee, T.-H.; Lee, J.-D. Phylogenetic analysis of the medicinal mushroom and taxonomical positions of their commercial products. J. Mushrooms 2005, 3, 52–59. [Google Scholar]
- Olennikov, D.N.; Sof’ya, V.A.; Tat’yana, A.P.; Borovski, G.B. Fatty acid composition of fourteen wood-decaying basidiomycete species growing in permafrost conditions. Rec. Nat. Prod. 2014, 8, 184. [Google Scholar]
- Ayoub, N.; Lass, D.; Schultze, W. Volatile constituents of the medicinal fungus chaga Inonotus obliquus (Pers.: Fr.) Pilát (Aphyllophoromycetideae). Int. J. Med. Mushrooms 2009, 11, 55–60. [Google Scholar] [CrossRef]
- Dimitrijevic, M.V.; Mitic, V.D.; Jovanovic, O.P.; Stankov Jovanovic, V.P.; Nikolic, J.S.; Petrovic, G.M.; Stojanovic, G.S. Comparative study of fatty acids profile in eleven wild mushrooms of Boletacea and Russulaceae families. Chem. Biodivers. 2018, 15, e1700434. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Molecular Formula | Precursor Ion [M-H]− | ppm Error | Fragment Ions [M-H]− by MS/MS a |
|---|---|---|---|---|
| 1.1 | C18H32O3 | 295.2278 (calcd. 295.2273) | −0.50 | 277.2176, 230.9864, 195.1398, 157.0871, 59.0144 |
| 1.2 | 295.2277 (calcd. 295.2273) | −0.82 | 277.2172, 211.1341, 183.1390, 111.0814 | |
| 1.3 | 295.2278 (calcd. 295.2273) | −0.63 | 277.2171, 195.1389, 183.1026, 155.1077, 59.0140 | |
| 1.4 | 295.2277 (calcd. 295.2273) | 1.33 | 277.2171, 195.1394, 171.1025 | |
| 2 | C18H30O3 | 293.2120 (calcd. 293.2117) | −0.57 | 249.2221, 195.1387, 167.1074, 113.0971, 59.0143 |
| 3 | C18H30O3 | 293.2120 (calcd. 293.2117) | −0.09 | 249.2220, 195.1388, 179.1075, 167.1075, 113.0971, 59.0141 |
| 4 | C18H30O3 | 293.2120 (calcd. 293.2117) | −0.51 | 249.2222, 221.1542, 197.1176, 185.1181, 125.0969 |
| 5 | C18H34O3 | 297.2446 (calcd. 297.2430) | −0.15 | 279.2327, 155.1077, 141.1284, 59.0142 |
| 6 | C18H32O2 | 279.2334 (calcd. 279.2324) | 1.64 | 233.1544, 178.8147, 134.8652, 96.9602, 83.0252, 68.9957 |
| 7 | C18H34O2 | 281.2488 (calcd. 281.2481) | 0.69 | 136.8628, 100.9336, 83.0251, 68.9965 |
| Linoleic acid | C18H32O2 | 279.2329 (calcd. 279.2324) | 1.64 | 233.1546, 178.8147, 134.8651, 96.9600, 83.0254, 68.9965 |
| Oleic acid | C18H34O2 | 281.2488 (calcd. 281.2481) | 0.69 | 136.8625, 100.9335, 83.0254, 68.9957 |
| Fatty Acids | F. officinalis | E. tinctorium | A. flettii | O. tomentosa |
|---|---|---|---|---|
| C11:1 | 0.0 | 0.0 | 0.2 | 0.0 |
| C12:0 | 0.8 | 1.0 | 0.2 | 0.3 |
| C12:1 | 0.5 | 0.5 | 0.0 | 0.1 |
| C14:0 | 0.3 | 0.5 | 0.4 | 0.5 |
| C14:1 | 0.0 | 0.6 | 0.1 | 0.2 |
| C15:0 | 0.3 | 2.0 | 2.1 | 1.5 |
| C15:1 | 0.7 | 0.7 | 0.0 | 0.2 |
| C16:0 | 6.0 | 14.6 | 8.1 | 14.1 |
| C16:1 | 0.3 | 2.1 | 0.9 | 0.7 |
| C16:1-trans | 0.3 | 0.9 | 0.3 | 0.2 |
| C17:0 | 0.4 | 0.8 | 2.2 | 0.3 |
| C17:1 | 0.0 | 1.1 | 0.0 | 0.0 |
| C18:0 | 4.5 | 10.4 | 3.7 | 4.3 |
| C18:1 (oleic) | 57.1 | 34.7 | 22.8 | 23.6 |
| C18:1 (vaccenic) | 2.6 | 2.8 | 2.2 | 1.5 |
| C18:2n6 (linoleic) | 17.1 | 15.9 | 51.7 | 48.4 |
| C18:2n6-trans | 0.0 | 0.5 | 0.1 | 0.1 |
| C18:3n3 | 0.0 | 0.1 | 0.2 | 0.2 |
| C18:3n6 | 0.4 | 0.5 | 0.1 | 0.1 |
| C18:4n3 | 0.4 | 0.3 | 0.0 | 0.0 |
| C19:1 | 0.0 | 0.2 | 0.1 | 0.0 |
| C20:0 | 0.3 | 0.9 | 0.4 | 0.3 |
| C20:1n11-cis | 0.4 | 0.6 | 0.2 | 0.3 |
| C20:1n15-cis | 3.4 | 1.2 | 0.2 | 0.2 |
| C20:2n6 | 0.3 | 0.4 | 1.1 | 0.3 |
| C20:3n6 | 1.0 | 0.9 | 0.1 | 0.2 |
| C20:4n6 | 0.3 | 1.4 | 0.1 | 0.2 |
| C20:5n3 | 0.0 | 0.3 | 0.1 | 0.3 |
| C22:0 | 0.4 | 1.1 | 0.9 | 0.3 |
| C22:1 | 0.3 | 0.2 | 0.3 | 0.1 |
| C22:4n6 | 0.3 | 0.0 | 0.0 | 0.0 |
| C22:5n6 | 0.6 | 0.0 | 0.0 | 0.0 |
| C22:6n3 | 0.7 | 2.3 | 0.6 | 0.4 |
| C24:0 | 0.2 | 0.5 | 0.1 | 0.1 |
| C24:1 | 0.0 | 0.0 | 0.2 | 0.8 |
| SFAs | 13.2 | 31.9 | 18.1 | 21.7 |
| UFAs | 86.8 | 68.1 | 81.9 | 78.3 |
| MUFAs | 65.5 | 45.7 | 27.6 | 27.9 |
| PUFAs | 21.2 | 22.4 | 54.3 | 50.5 |
| Omega-3 Fatty Acids | 1.2 | 3.0 | 0.9 | 1.0 |
| Omega-6 Fatty Acids | 20.0 | 19.4 | 53.4 | 49.5 |
| trans Fatty Acids | 0.3 | 1.4 | 0.3 | 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.X.; Li, W.M.; Khatra, J.; Xia, Z.; Sannikov, O.; Ling, Y.; Zhu, H.; Lee, C.H. Antiproliferative Fatty Acids Isolated from the Polypore Fungus Onnia tomentosa. J. Fungi 2022, 8, 1163. https://doi.org/10.3390/jof8111163
Lee HX, Li WM, Khatra J, Xia Z, Sannikov O, Ling Y, Zhu H, Lee CH. Antiproliferative Fatty Acids Isolated from the Polypore Fungus Onnia tomentosa. Journal of Fungi. 2022; 8(11):1163. https://doi.org/10.3390/jof8111163
Chicago/Turabian StyleLee, Hooi Xian, Wai Ming Li, Jatinder Khatra, Zhicheng Xia, Oleg Sannikov, Yun Ling, Haoxuan Zhu, and Chow H. Lee. 2022. "Antiproliferative Fatty Acids Isolated from the Polypore Fungus Onnia tomentosa" Journal of Fungi 8, no. 11: 1163. https://doi.org/10.3390/jof8111163