
Accounting for the Biological Complexity of Pathogenic Fungi in Phylogenetic Dating


Abstract
:1. Introduction
2. Methodological Considerations for Temporal Analyses of Pathogenic Fungi
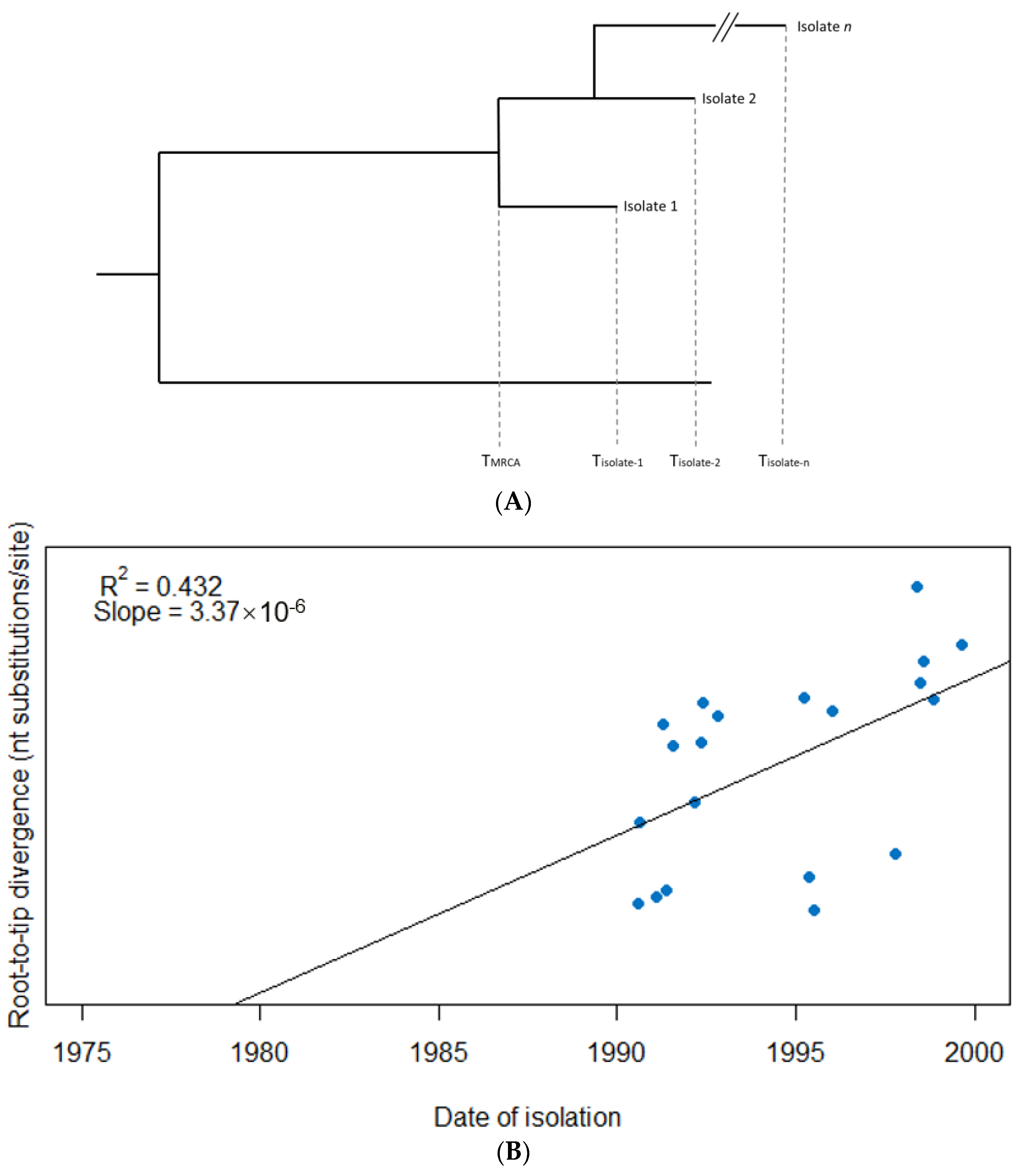
2.1. Detection of a Temporal Signal in Sequence Data
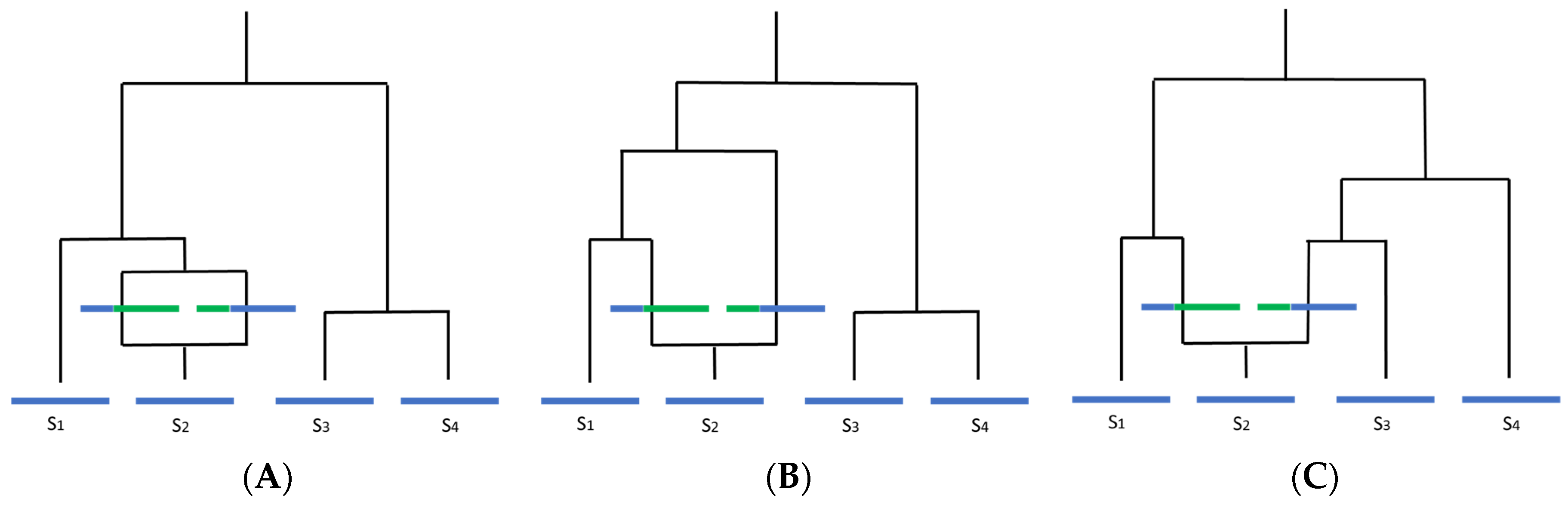
2.2. Recombination Needs to Be Considered and Accounted for
2.3. Quiescence to Hypermutation—Potential Impact of Mutation Rate Variation
2.4. Potential Sensitivity to Priors Requires Explicit Sharing of Bayesian Model Settings
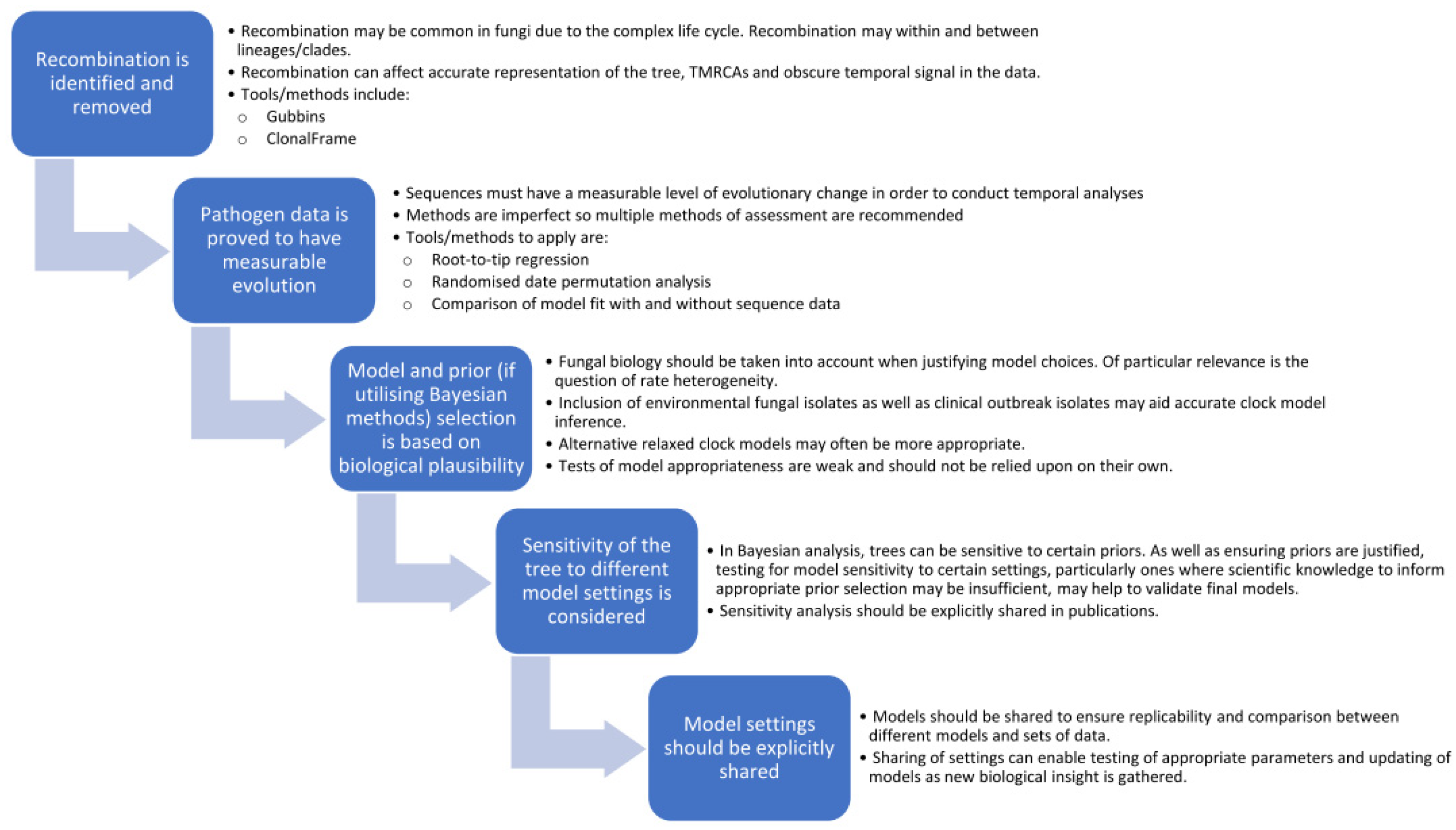
3. Recommendations for the Way Forward
- Identify and remove regions of recombination, including recombination both within and between sub-populations/clades. This can be done using freely available tools, such as Gubbins or ClonalFrameML, and should be conducted first as recombination can hinder detection of a temporal signal in the data.
- Test data for temporal signal through a combination of methods including root-to-tip regression, data randomisation and improved model fit. Importantly, if the data does not have measurable temporal signals, then dating analyses should not be applied.
- Consider the biology of the organism and potential rate heterogeneity in the selection of appropriate clock models, particularly if datasets are limited to just clinical outbreak isolates or if common ancestors are far back in time. Tests of model fit between different clock (and demographic) models can have poor performance and should not be solely relied upon [108]. Since most fungal infections are saprophytic, stemming from the environment, inclusion of environmental fungal isolates in a phylogeny may provide better indication of heterogeneity in clock rates. Further research into fungal states of hypermutation and quiescence may shed more light on these variations and how frequent or infrequent they may be.
- If utilising Bayesian analysis, there should be a biological justification for prior settings, and all should be explicitly shared in publications. This can ensure that models are replicable and would allow for comparison between different models. Since final trees can be affected by sensitivity to certain priors, being explicit with model settings would allow for this to be investigated. Studies could examine the sensitivity of the final tree to clock rate and tree priors to justify (or question) the validity of the final tree.
Author Contributions
Funding
Conflicts of Interest
References
- Grenfell, B.T.; Pybus, O.; Gog, J.; Wood, J.L.N.; Daly, J.; Mumford, J.A.; Holmes, E. Unifying the Epidemiological and Evolutionary Dynamics of Pathogens. Science 2004, 303, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Volz, E.M.; Koelle, K.; Bedford, T. Viral Phylodynamics. PLoS Comput. Biol. 2013, 9, e1002947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Hedges, S.B. Advances in Time Estimation Methods for Molecular Data. Mol. Biol. Evol. 2016, 33, 863–869. [Google Scholar] [CrossRef] [Green Version]
- Boskova, V.; Bonhoeffer, S.; Stadler, T. Inference of Epidemiological Dynamics Based on Simulated Phylogenies Using Birth-Death and Coalescent Models. PLoS Comput. Biol. 2014, 10, e1003913. [Google Scholar] [CrossRef] [Green Version]
- Pybus, O.G.; Rambaut, A. Evolutionary analysis of the dynamics of viral infectious disease. Nat. Rev. Genet. 2009, 10, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C.; Dudas, G.; Rambaut, A.; Andersen, K.G. The evolution of Ebola virus: Insights from the 2013–2016 epidemic. Nature 2016, 538, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Barzilai, L.P.; Schrago, C.G. The range of sampling times affects Zika virus evolutionary rates and divergence times. Arch. Virol. 2019, 164, 3027–3034. [Google Scholar] [CrossRef]
- Duchene, S.; Holt, K.; Weill, F.-X.; Le Hello, S.; Hawkey, J.; Edwards, D.J.; Fourment, M.; Holmes, E.C. Genome-scale rates of evolutionary change in bacteria. Microb. Genom. 2016, 2, e000094. [Google Scholar] [CrossRef]
- Walker, T.M.; Ip, C.L.; Harrell, R.H.; Evans, J.T.; Kapatai, G.; Dedicoat, M.J.; Eyre, D.; Wilson, D.; Hawkey, P.M.; Crook, D.W.; et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet Infect. Dis. 2013, 13, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Roe, C.C.; Bowers, J.; Oltean, H.; DeBess, E.; Dufresne, P.J.; McBurney, S.; Overy, D.P.; Wanke, B.; Lysen, C.; Chiller, T.; et al. Dating the Cryptococcus gattii Dispersal to the North American Pacific Northwest. mSphere 2018, 3, e00499-17. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, J.; Farrer, R.A.; Desjardins, C.A.; Gallo, J.E.; Sykes, S.; Sakthikumar, S.; Misas, E.; Whiston, E.A.; Bagagli, E.; Soares, C.M.A.; et al. Genome Diversity, Recombination, and Virulence across the Major Lineages of Paracoccidioides. mSphere 2016, 1, e00213-16. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, J.; Abdolrasouli, A.; Farrer, R.A.; Cuomo, C.A.; Aanensen, D.M.; Armstrong-James, D.; Fisher, M.C.; Schelenz, S. Genomic Epidemiology of the UK Outbreak of the Emerging Human Fungal Pathogen Candida auris. Emerg. Microbes Infect. 2018, 7, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, N.A.; Muñoz, J.F.; Gade, L.; Berkow, E.L.; Li, X.; Welsh, R.M.; Forsberg, K.; Lockhart, S.R.; Adam, R.; Alanio, A.; et al. Tracing the Evolutionary History and Global Expansion of Candida auris Using Population Genomic Analyses. mBio 2020, 11, 03364-19. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.C.; Henk, D.A.; Briggs, C.J.; Brownstein, J.S.; Madoff, L.C.; McCraw, S.L.; Gurr, S.J. Emerging fungal threats to animal, plant and ecosystem health. Nature 2012, 484, 186–194. [Google Scholar] [CrossRef]
- Garcia-Solache, M.A.; Casadevall, A. Hypothesis: Global Warming Will Bring New Fungal Diseases for Mammals. mBio 2010, 1, 00061-10. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.C.; Gurr, S.J.; Cuomo, C.A.; Blehert, D.S.; Jin, H.; Stukenbrock, E.H.; Stajich, J.E.; Kahmann, R.; Boone, C.; Denning, D.W.; et al. Threats Posed by the Fungal Kingdom to Humans, Wildlife, and Agriculture. mBio 2020, 11, 00449-20. [Google Scholar] [CrossRef]
- Benedict, K.; Richardson, M.; Vallabhaneni, S.; Jackson, B.R.; Chiller, T. Emerging issues, challenges, and changing epidemiology of fungal disease outbreaks. Lancet Infect. Dis. 2017, 17, e403–e411. [Google Scholar] [CrossRef]
- Bougnoux, M.-E.; Brun, S.; Zahar, J.-R. Healthcare-associated fungal outbreaks: New and uncommon species, New molecular tools for investigation and prevention. Antimicrob. Resist. Infect. Control. 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, T.; Kouyos, R.; von Wyl, V.; Yerly, S.; Böni, J.; Bürgisser, P.; Klimkait, T.; Joos, B.; Rieder, P.; Xie, D.; et al. Estimating the Basic Reproductive Number from Viral Sequence Data. Mol. Biol. Evol. 2012, 29, 347–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, F.; Hughes, G.J.; Rambaut, A.; Pozniak, A.; Brown, A.J.L. Episodic Sexual Transmission of HIV Revealed by Molecular Phylodynamics. PLoS Med. 2008, 5, e50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, T.; Comas, I.; Luo, D.; Lu, B.; Wu, J.; Wei, L.; Yang, C.; Liu, Q.; Gan, M.; Sun, G.; et al. Southern East Asian origin and coexpansion ofMycobacterium tuberculosisBeijing family with Han Chinese. Proc. Natl. Acad. Sci. USA 2015, 112, 8136–8141. [Google Scholar] [CrossRef] [Green Version]
- Eldholm, V.; Pettersson, J.H.-O.; Brynildsrud, O.; Kitchen, A.; Rasmussen, E.M.; Lillebaek, T.; Rønning, J.O.; Crudu, V.; Mengshoel, A.T.; Debech, N.; et al. Armed conflict and population displacement as drivers of the evolution and dispersal of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2016, 113, 13881–13886. [Google Scholar] [CrossRef] [Green Version]
- Merker, M.; Blin, C.; Mona, S.; Duforet-Frebourg, N.; Lecher, S.; Willery, E.; Blum, M.; Rüsch-Gerdes, S.; Mokrousov, I.; Aleksic, E.; et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat. Genet. 2015, 47, 242–249. [Google Scholar] [CrossRef]
- Dudas, G.; Carvalho, L.M.; Bedford, T.; Tatem, A.J.; Baele, G.; Faria, N.R.; Park, D.J.; Ladner, J.T.; Arias, A.; Asogun, D.; et al. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 2017, 544, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Dellicour, S.; Baele, G.; Dudas, G.; Faria, N.R.; Pybus, O.; Suchard, M.A.; Rambaut, A.; Lemey, P. Phylodynamic assessment of intervention strategies for the West African Ebola virus outbreak. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Borman, A.M.; Johnson, E.M. Candida auris in the UK: Introduction, dissemination, and control. PLOS Pathog. 2020, 16, e1008563. [Google Scholar] [CrossRef]
- Schelenz, S.; Hagen, F.; Rhodes, J.L.; Abdolrasouli, A.; Chowdhary, A.; Hall, A.; Ryan, L.; Shackleton, J.; Trimlett, R.; Meis, J.F.; et al. First hospital outbreak of the globally emerging Candida auris in a European hospital. Antimicrob. Resist. Infect. Control. 2016, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.G.; Shin, J.H.; Uh, Y.; Kang, M.G.; Kim, S.H.; Park, K.H.; Jang, H.-C. First Three Reported Cases of Nosocomial Fungemia Caused by Candida auris. J. Clin. Microbiol. 2011, 49, 3139–3142. [Google Scholar] [CrossRef] [Green Version]
- Toyotome, T.; Hagiwara, D.; Takahashi, H.; Watanabe, A.; Kamei, K. Emerging Antifungal Drug Resistance in Aspergillus fumigatus and Among Other Species of Aspergillus. Curr. Fungal Infect. Rep. 2018, 12, 105–111. [Google Scholar] [CrossRef]
- Worku, M.; Girma, F. Candida auris: From Multidrug Resistance to Pan-Resistant Strains. Infect. Drug Resist. 2020, 13, 1287–1294. [Google Scholar] [CrossRef]
- Du, H.; Bing, J.; Hu, T.; Ennis, C.L.; Nobile, C.J.; Huang, G. Candida auris: Epidemiology, biology, antifungal resistance, and virulence. PLOS Pathog. 2020, 16, e1008921. [Google Scholar] [CrossRef]
- Smith, K.D.; Achan, B.; Hullsiek, K.H.; McDonald, T.R.; Okagaki, L.H.; Alhadab, A.A.; Akampurira, A.; Rhein, J.; Meya, D.B.; Boulware, D.; et al. Increased Antifungal Drug Resistance in Clinical Isolates of Cryptococcus neoformans in Uganda. Antimicrob. Agents Chemother. 2015, 59, 7197–7204. [Google Scholar] [CrossRef] [Green Version]
- Zafar, H.; Altamirano, S.; Ballou, E.R.; Nielsen, K. A titanic drug resistance threat in Cryptococcus neoformans. Curr. Opin. Microbiol. 2019, 52, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Emergence of Cryptococcus gattii—Pacific Northwest, 2004–2010. MMWR 2010, 59, 865–868. [Google Scholar]
- Bartlett, K.H.; Kidd, S.; Kronstad, J.W. The emergence of Cryptococcus gattii in British Columbia and the Pacific Northwest. Curr. Infect. Dis. Rep. 2008, 10, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, 01031-17. [Google Scholar] [CrossRef] [Green Version]
- Menardo, F.; Duchêne, S.; Brites, D.; Gagneux, S. The molecular clock of Mycobacterium tuberculosis. PLOS Pathog. 2019, 15, e1008067. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.; Shackelton, L.A.; Holmes, E. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.; Pybus, O.; Rambaut, A. Inference of Viral Evolutionary Rates from Molecular Sequences. Adv. Parasitol. 2003, 54, 331–358. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Murray, G.; Wang, F.; Harrison, E.M.; Paterson, G.; Mather, A.E.; Harris, S.R.; Holmes, M.A.; Rambaut, A.; Welch, J.J. The effect of genetic structure on molecular dating and tests for temporal signal. Methods Ecol. Evol. 2015, 7, 80–89. [Google Scholar] [CrossRef]
- Didelot, X.; Croucher, N.; Bentley, S.D.; Harris, S.R.; Wilson, D. Bayesian inference of ancestral dates on bacterial phylogenetic trees. Nucleic Acids Res. 2018, 46, e134. [Google Scholar] [CrossRef] [Green Version]
- Ramsden, C.; Melo, F.L.; Figueiredo, L.M.; Holmes, E.C.; Zanotto, P.M.A. High rates of molecular evolution in hantaviruses. Mol. Biol. Evol. 2008, 25, 1488–1492. [Google Scholar] [CrossRef] [Green Version]
- Duchene, S.; Duchêne, D.A.; Holmes, E.; Ho, S.Y. The Performance of the Date-Randomization Test in Phylogenetic Analyses of Time-Structured Virus Data. Mol. Biol. Evol. 2015, 32, 1895–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ene, I.V.; Farrer, R.; Hirakawa, M.; Agwamba, K.; Cuomo, C.A.; Bennett, R.J. Global analysis of mutations driving microevolution of a heterozygous diploid fungal pathogen. Proc. Natl. Acad. Sci. USA 2018, 115, E8688–E8697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.O.; Siegal, M.; Hall, D.; Petrov, D.A. Precise estimates of mutation rate and spectrum in yeast. Proc. Natl. Acad. Sci. USA 2014, 111, E2310–E2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelthaler, D.M.; Casadevall, A. On the Emergence of Cryptococcus gattii in the Pacific Northwest: Ballast Tanks, Tsunamis, and Black Swans. mBio 2019, 10, e02193-19. [Google Scholar] [CrossRef] [Green Version]
- Duina, A.A.; Miller, M.E.; Keeney, J.B. Budding Yeast for Budding Geneticists: A Primer on the Saccharomyces cerevisiae Model System. Genetics 2014, 197, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Tavanti, A.; Gow, N.A.; Maiden, M.C.; Odds, F.C.; Shaw, D.J. Genetic evidence for recombination in Candida albicans based on haplotype analysis. Fungal Genet. Biol. 2004, 41, 553–562. [Google Scholar] [CrossRef]
- Billmyre, R.B.; Croll, D.; Li, W.; Mieczkowski, P.; Carter, D.A.; Cuomo, C.; Kronstad, J.W.; Heitman, J. Highly Recombinant VGII Cryptococcus gattii Population Develops Clonal Outbreak Clusters through both Sexual Macroevolution and Asexual Microevolution. mBio 2014, 5, e01494-14. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Patel, S.M.; Litvintseva, A.P.; Floyd, A.; Mitchell, T.G.; Heitman, J. Diploids in the Cryptococcus neoformans Serotype A Population Homozygous for the α Mating Type Originate via Unisexual Mating. PLOS Pathog. 2009, 5, e1000283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon-Chung, K.J.; Bennett, J.E. Distribution of α and α mating types of Cryptococcus neoformans among natural and clinical isolates. Am. J. Epidemiol. 1978, 108, 337–340. [Google Scholar] [CrossRef]
- Litvintseva, A.P.; Marra, R.E.; Nielsen, K.; Heitman, J.; Vilgalys, R.; Mitchell, T.G. Evidence of Sexual Recombination among Cryptococcus neoformans Serotype A Isolates in Sub-Saharan Africa. Eukaryot. Cell 2003, 2, 1162–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, R.J. Coming of Age—Sexual Reproduction in Candida Species. PLOS Pathog. 2010, 6, e1001155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.G.; Johnson, A.D. White-Opaque Switching in Candida albicans Is Controlled by Mating-Type Locus Homeodomain Proteins and Allows Efficient Mating. Cell 2002, 110, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Roth, C.; Sun, S.; Billmyre, R.B.; Heitman, J.; Magwene, P.M. A High-Resolution Map of Meiotic Recombination in Cryptococcus deneoformans Demonstrates Decreased Recombination in Unisexual Reproduction. Genetics 2018, 209, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Gunge, N.; Nakatomi, Y. Genetic Mechanisms of Rare Matings of the Yeast Saccharomyces cerevisiae Heterozygous for Mating Type. Genetics 1972, 70, 41–58. [Google Scholar] [CrossRef]
- Anderson, M.Z.; Thomson, G.J.; Hirakawa, M.P.; Bennett, R.J. A ‘parameiosis’ drives depolyploidization and homologous recombination in Candida albicans. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Schierup, M.H.; Hein, J. Consequences of Recombination on Traditional Phylogenetic Analysis. Genetics 2000, 156, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Hein, J.; Schierup, M.; Wiuf, C. Gene Genealogies, Variation and Evolution: A Primer in Coalescent Theory; Oxford University Press: Oxford, UK; New York, NY, USA, 2005. [Google Scholar]
- Anisimova, M.; Nielsen, R.; Yang, Z. Effect of Recombination on the Accuracy of the Likelihood Method for Detecting Positive Selection at Amino Acid Sites. Genetics 2003, 164, 1229–1236. [Google Scholar] [CrossRef]
- Shriner, D.; Nickle, D.C.; Jensen, M.; Mullins, J.I. Potential impact of recombination on sitewise approaches for detecting positive natural selection. Genet. Res. 2003, 81, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Posada, D.; Crandall, K.A. The Effect of Recombination on the Accuracy of Phylogeny Estimation. J. Mol. Evol. 2002, 54, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Hedge, J.; Wilson, D.J. Bacterial Phylogenetic Reconstruction from Whole Genomes Is Robust to Recombination but Demographic Inference Is Not. mBio 2014, 5, e02158-14. [Google Scholar] [CrossRef] [Green Version]
- Arenas, M.; Posada, D. The Effect of Recombination on the Reconstruction of Ancestral Sequences. Genetics 2010, 184, 1133–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015, 43, e15. [Google Scholar] [CrossRef] [Green Version]
- Didelot, X.; Wilson, D.J. ClonalFrameML: Efficient Inference of Recombination in Whole Bacterial Genomes. PLoS Comput. Biol. 2015, 11, e1004041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostowy, R.; Croucher, N.J.; Andam, C.P.; Corander, J.; Hanage, W.P.; Marttinen, P. Efficient Inference of Recent and Ancestral Recombination within Bacterial Populations. Mol. Biol. Evol. 2017, 34, 1167–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeois, Y.X.C. Going down the rabbit hole: A review on how to link genome-wide data with ecology and evolution in natural populations. bioRxiv 2016, 052761. [Google Scholar] [CrossRef]
- Billmyre, R.B.; Clancey, S.A.; Li, L.X.; Doering, T.L.; Heitman, J. 5-fluorocytosine resistance is associated with hypermutation and alterations in capsule biosynthesis in Cryptococcus. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billmyre, R.B.; Clancey, S.A.; Li, L.X.; Doering, T.L.; Heitman, J. Hypermutation in Cryptococcus Reveals a Novel Pathway to 5-Fluorocytosine (5FC) Resistance. bioRxiv 2019, 636928. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, J.; Beale, M.A.; Vanhove, M.; Jarvis, J.N.; Kannambath, S.; Simpson, J.A.; Ryan, A.; Meintjes, G.; Harrison, T.; Fisher, M.C.; et al. A Population Genomics Approach to Assessing the Genetic Basis of Within-Host Microevolution Underlying Recurrent Cryptococcal Meningitis Infection. G3 Genes Genomes Genet. 2017, 7, 1165–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weil, T.; Santamaría, R.; Lee, W.; Rung, J.; Tocci, N.; Abbey, D.; Bezerra, A.; Carreto, L.; Moura, G.R.; Bayés, M.; et al. Adaptive Mistranslation Accelerates the Evolution of Fluconazole Resistance and Induces Major Genomic and Gene Expression Alterations in Candida albicans. mSphere 2017, 2, 00167-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avramovska, O.; Hickman, M.A. The Magnitude of Candida albicans Stress-Induced Genome Instability Results from an Interaction Between Ploidy and Antifungal Drugs. G3 Genes Genomes Genet. 2019, 9, 4019–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeClerc, J.E.; Li, B.; Payne, W.L.; Cebula, T.A. High Mutation Frequencies among Escherichia coli and Salmonella Pathogens. Sci. Science 1996, 274, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, S.; Desbordes, L.; Gracieux, P.; Saffroy, S.; Bousarghin, L.; Bonnaure-Mallet, M.; Jolivet-Gougeon, A. Distribution of mutation frequencies among Salmonella enterica isolates from animal and human sources and genetic characterization of a Salmonella Heidelberg hypermutator. Vet. Microbiol. 2009, 137, 306–312. [Google Scholar] [CrossRef]
- Sagot, I.; Laporte, D. The cell biology of quiescent yeast—A diversity of individual scenarios. J. Cell Sci. 2019, 132, jcs213025. [Google Scholar] [CrossRef] [Green Version]
- Salyer, W.R.; Salyer, D.C.; Baker, R.D. Primary Complex of Cryptococcus and Pulmonary Lymph Nodes. J. Infect. Dis. 1974, 130, 74–77. [Google Scholar] [CrossRef]
- Goldman, D.L.; Lee, S.C.; Mednick, A.J.; Montella, L.; Casadevall, A. Persistent Cryptococcus neoformans Pulmonary Infection in the Rat Is Associated with Intracellular Parasitism, Decreased Inducible Nitric Oxide Synthase Expression, and Altered Antibody Responsiveness to Cryptococcal Polysaccharide. Infect. Immun. 2000, 68, 832–838. [Google Scholar] [CrossRef] [Green Version]
- MacDougall, L.; Fyfe, M. Emergence of Cryptococcus gattii in a Novel Environment Provides Clues to Its Incubation Period. J. Clin. Microbiol. 2006, 44, 1851–1852. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Hermoso, D.; Janbon, G.; Dromer, F. Epidemiological Evidence for Dormant Cryptococcus neoformans Infection. J. Clin. Microbiol. 1999, 37, 3204–3209. [Google Scholar] [CrossRef] [Green Version]
- Castro-Lainez, M.T.; Sierra-Hoffman, M.; Llompart-Zeno, J.; Adams, R.; Howell, A.; Hoffman-Roberts, H.; Fader, R.; Arroliga, A.C.; Jinadatha, C. Talaromyces marneffei infection in a non-HIV non-endemic population. IDCases 2018, 12, 21–24. [Google Scholar] [CrossRef]
- Dutta, N.K.; Karakousis, P.C. Latent Tuberculosis Infection: Myths, Models, and Molecular Mechanisms. Microbiol. Mol. Biol. Rev. 2014, 78, 343–371. [Google Scholar] [CrossRef] [Green Version]
- Ford, C.B.; Lin, P.L.; Chase, M.R.; Shah, R.R.; Iartchouk, O.; Galagan, J.; Mohaideen, N.; Ioerger, T.R.; Sacchettini, J.C.; Lipsitch, M.; et al. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat. Genet. 2011, 43, 482–486. [Google Scholar] [CrossRef] [Green Version]
- Lillebaek, T.; Dirksen, A.; Baess, I.; Strunge, B.; Thomsen, V.Ø.; Andersen, Å.B. Molecular Evidence of Endogenous Reactivation ofMycobacterium tuberculosisafter 33 Years of Latent Infection. J. Infect. Dis. 2002, 185, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Lillebaek, T.; Dirksen, A.; Vynnycky, E.; Baess, I.; Thomsen, V.Ø.; Andersen, Å.B. Stability of DNA Patterns and Evidence ofMycobacterium tuberculosisReactivation Occurring Decades after the Initial Infection. J. Infect. Dis. 2003, 188, 1032–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colangeli, R.; Arcus, V.L.; Cursons, R.T.; Ruthe, A.; Karalus, N.; Coley, K.; Manning, S.; Kim, S.; Marchiano, E.; Alland, D. Whole Genome Sequencing of Mycobacterium tuberculosis Reveals Slow Growth and Low Mutation Rates during Latent Infections in Humans. PLoS ONE 2014, 9, e91024. [Google Scholar] [CrossRef]
- Colangeli, R.; Gupta, A.; Vinhas, S.A.; Venkata, U.D.C.; Kim, S.; Grady, C.; Jones-López, E.C.; Soteropoulos, P.; Palaci, M.; Marques-Rodrigues, P.; et al. Mycobacterium tuberculosis progresses through two phases of latent infection in humans. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Roche, B.; Arcangioli, B.; Martienssen, R. Transcriptional reprogramming in cellular quiescence. RNA Biol. 2017, 14, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Gray, J.V.; Petsko, G.A.; Johnston, G.C.; Ringe, D.; Singer, R.A.; Werner-Washburne, M. “Sleeping Beauty”: Quiescence in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2004, 68, 187–206. [Google Scholar] [CrossRef] [Green Version]
- De Virgilio, C. The essence of yeast quiescence. FEMS Microbiol. Rev. 2012, 36, 306–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rittershaus, E.S.; Baek, S.-H.; Sassetti, C.M. The Normalcy of Dormancy: Common Themes in Microbial Quiescence. Cell Host Microbe 2013, 13, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Ashton, P.M.; Thanh, L.T.; Trieu, P.H.; Van Anh, D.; Trinh, N.M.; Beardsley, J.; Kibengo, F.; Chierakul, W.; Dance, D.A.B.; Rattanavong, S.; et al. Three phylogenetic groups have driven the recent population expansion of Cryptococcus neoformans. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gangloff, S.; Achaz, G.; Francesconi, S.; Villain, A.; Miled, S.; Denis, C.; Arcangioli, B. Quiescence unveils a novel mutational force in fission yeast. eLife 2017, 6, e27469. [Google Scholar] [CrossRef]
- Biek, R.; Pybus, O.; Lloyd-Smith, J.O.; Didelot, X. Measurably evolving pathogens in the genomic era. Trends Ecol. Evol. 2015, 30, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Thorne, J.L.; Kishino, H.; Painter, I. Estimating the rate of evolution of the rate of molecular evolution. Mol. Biol. Evol. 1998, 15, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Larget, B.; Swofford, D. A Compound Poisson Process for Relaxing the Molecular Clock. Genetics 2000, 154, 1879–1892. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef] [PubMed]
- Yoder, A.D.; Yang, Z. Estimation of Primate Speciation Dates Using Local Molecular Clocks. Mol. Biol. Evol. 2000, 17, 1081–1090. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A. Bayesian random local clocks, or one rate to rule them all. BMC Biol. 2010, 8, 114. [Google Scholar] [CrossRef] [Green Version]
- Didelot, X.; Siveroni, I.; Volz, E.M. Additive Uncorrelated Relaxed Clock Models for the Dating of Genomic Epidemiology Phylogenies. Mol. Biol. Evol. 2021, 38, 307–317. [Google Scholar] [CrossRef]
- Drummond, A.J.; Bouckaert, R.R. BEAST: Bayesian Evolutionary Analysis by Sampling Trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Dos Reis, M.; Donoghue, P.; Yang, Z. Bayesian molecular clock dating of species divergences in the genomics era. Nat. Rev. Genet. 2016, 17, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, F.F.; Dos Reis, M.; Yang, Z. A biologist’s guide to Bayesian phylogenetic analysis. Nat. Ecol. Evol. 2017, 1, 1446–1454. [Google Scholar] [CrossRef] [Green Version]
- Möller, S.; du Plessis, L.; Stadler, T. Impact of the tree prior on estimating clock rates during epidemic outbreaks. Proc. Natl. Acad. Sci. USA 2018, 115, 4200–4205. [Google Scholar] [CrossRef] [Green Version]
- Angelis, K.; Álvarez-Carretero, S.; Dos Reis, M.; Yang, Z. An Evaluation of Different Partitioning Strategies for Bayesian Estimation of Species Divergence Times. Syst. Biol. 2018, 67, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Kainer, D.; Lanfear, R. The Effects of Partitioning on Phylogenetic Inference. Mol. Biol. Evol. 2015, 32, 1611–1627. [Google Scholar] [CrossRef] [Green Version]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Task | Tool/Algorithm | Description | Reference |
|---|---|---|---|
| Identify loci of recombination | Gubbins | Iteratively identifies loci of recombination and simultaneously constructs a phylogeny based on point mutations outside of these identified regions. | Croucher et al., Nucleic Acids Res. 2014, doi:10.1093/nar/gku1196 |
| ClonalFrameML | Maximum likelihood inference to detect loci of recombination and simultaneously construct a phylogeny accounting for this recombination. | Didelot and Wilson, PLoS Comput. Biol. 2015, doi:10.1371/journal.pcbi.10004041 | |
| fastGEAR | Identifies population genetic structure of an (species-wide) alignment and detects recombination, both recent and ancestral, between inferred lineages as well as recent recombination from external origins using a hidden Markov model. | Mostowy et al., Mol. Biol. Evol. 2017, doi:10.1093/molbev/msx066 | |
| Phylogenetic inference | BEAST/BEAST2 | Bayesian MCMC algorithm to jointly estimate a phylogeny and its associated parameters (i.e., effective population size, TMRCA, clock rate, etc.) | Drummond and Rambaut, BMC Evol. Biol. 2007, doi:10.1017/CBO9781139095112.007 |
| MrBayes | Bayesian MCMC inference and model choice across a range of phylogenetic and evolutionary models | Huelsenbeck and Ronquist, Bioinformatics. 2001, 17 (8), 754–755 | |
| IQ-Tree | Stochastic tree-searching algorithm to identify the highest likelihood tree (output in nucleotide substitutions only, not calendar time) | Nguyen et al., Mol. Biol. Evol. 2015, 32(1):268–74. | |
| PhyML | ML inference of phylogenetic relationships between divergent populations, utilising subtree pruning and regrafting (SPR) and approximate likelihood-ratio test (aLRT) approaches. | Guindon et al., Systematic Biol. 2010, 59 (3), 307–321. | |
| RAxML | ML inference of phylogenetic relationships between divergent populations utilising parsimony and heuristic subtree rearrangements. | Stamatakis, Bioinformatics. 2014, 30 (9), 1312–1313. | |
| PHYLIP | Package of programmes for phylogenetic inference including parsimony, distance matrix and ML methods, bootstrapping and consensus trees. | Felsenstein, Cladistics. 1989, 5 (2), 163–166. | |
| SNPhylo | Pipeline utilising ML to reconstruct phylogenies based on SNP data. | Lee et al., BMC Genomics. 2014, 15, 162. | |
| Molecular clock rate/Divergence time estimation | Treedater | R package to apply an evolutionary timescale to date and root a phylogeny (i.e., transforms branch lengths from number of nucleotide substitutions to calendar time) and estimate TMRCA. Molecular clock test function tests for appropriate clock model (relaxed vs. strict). | Volz and Frost, Virus Evol. 2017, doi:10.1093/ve/vex025 |
| PhyTime | A tool in the PhyML package that estimates divergence dates in a Bayesian setting. | Guindon, Systematic Biol. 2013, 62 (1), 22–34 Top of Form Bottom of Form | |
| Phylogeny viewer/editor | Figtree | Graphical viewer of phylogenetic trees and to produce publication-ready figures. Particularly suited to trees produced by BEAST. | http://tree.bio.ed.ac.uk/software/figtree/ accessed on 11 August 2021 |
| Icytree | A simple browser-based phylogenetic tree viewer. | https://icytree.org/ accessed on 11 August 2021 | |
| GGTREE | An R package for programmable visualisation and annotation of phylogenetic trees. | Guangchuang et al., Methods in Ecology and Evolution. 8 (1), 28–36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edwards, H.M.; Rhodes, J. Accounting for the Biological Complexity of Pathogenic Fungi in Phylogenetic Dating. J. Fungi 2021, 7, 661. https://doi.org/10.3390/jof7080661
Edwards HM, Rhodes J. Accounting for the Biological Complexity of Pathogenic Fungi in Phylogenetic Dating. Journal of Fungi. 2021; 7(8):661. https://doi.org/10.3390/jof7080661
Chicago/Turabian StyleEdwards, Hannah M., and Johanna Rhodes. 2021. "Accounting for the Biological Complexity of Pathogenic Fungi in Phylogenetic Dating" Journal of Fungi 7, no. 8: 661. https://doi.org/10.3390/jof7080661