
Genomic Diversity Analysis Reveals a Strong Population Structure in Histoplasma capsulatum LAmA (Histoplasma suramericanum)

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Isolation and Culture Conditions
2.2. Patient’s Information
2.3. DNA Extraction
2.4. Whole Genome Sequencing and SNP Variant Calling
2.5. Phylogenetic Trees Using Whole Genomes
2.6. Population Structure and Admixture
2.7. Clinical Differences between RJ and Non-RJ Samples
2.8. Strain Genotyping Using Multi Locus Sequencing Type (MLST) Analysis
3. Results
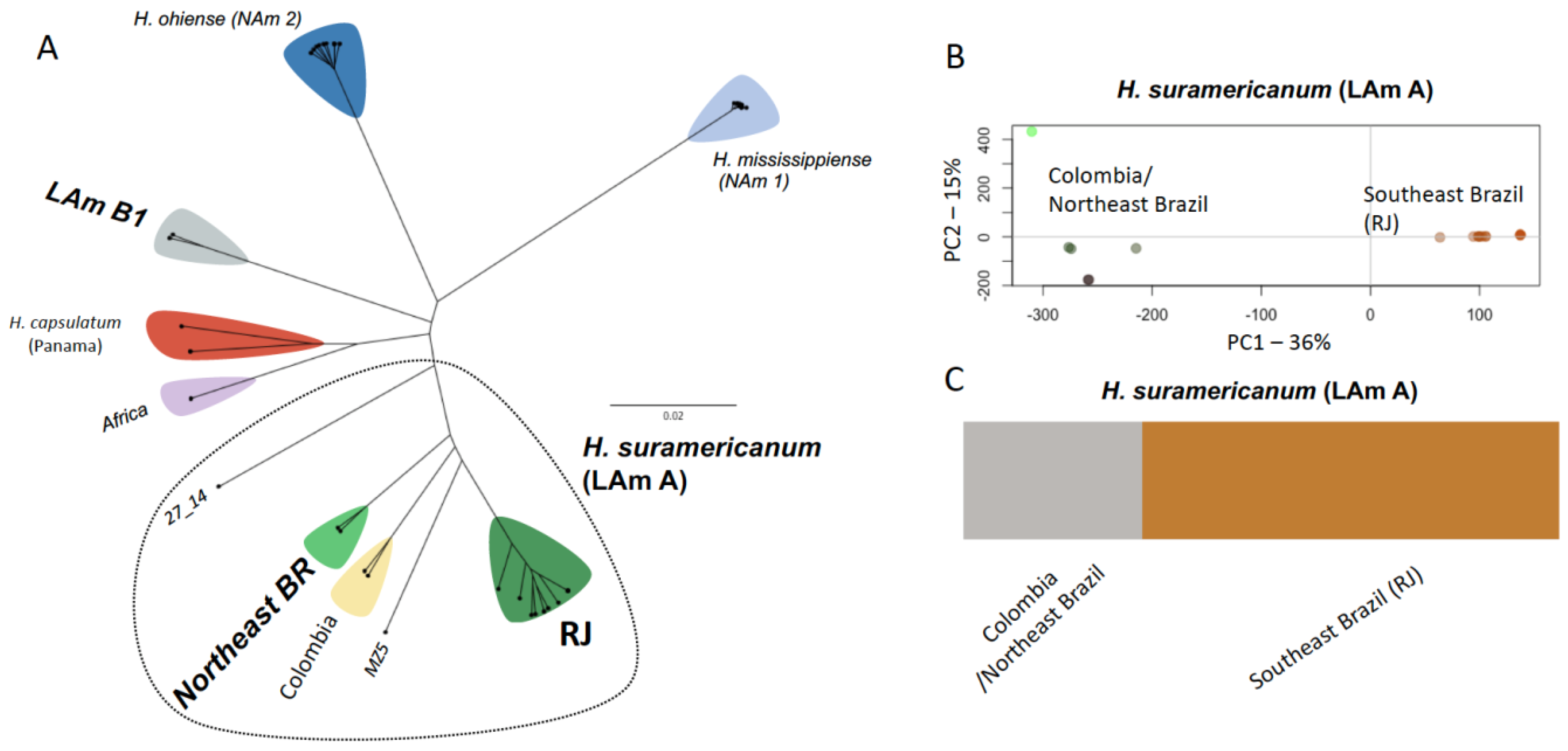
3.1. Phylogenomic Diversity
3.2. Population Structure within H. Suramericanum
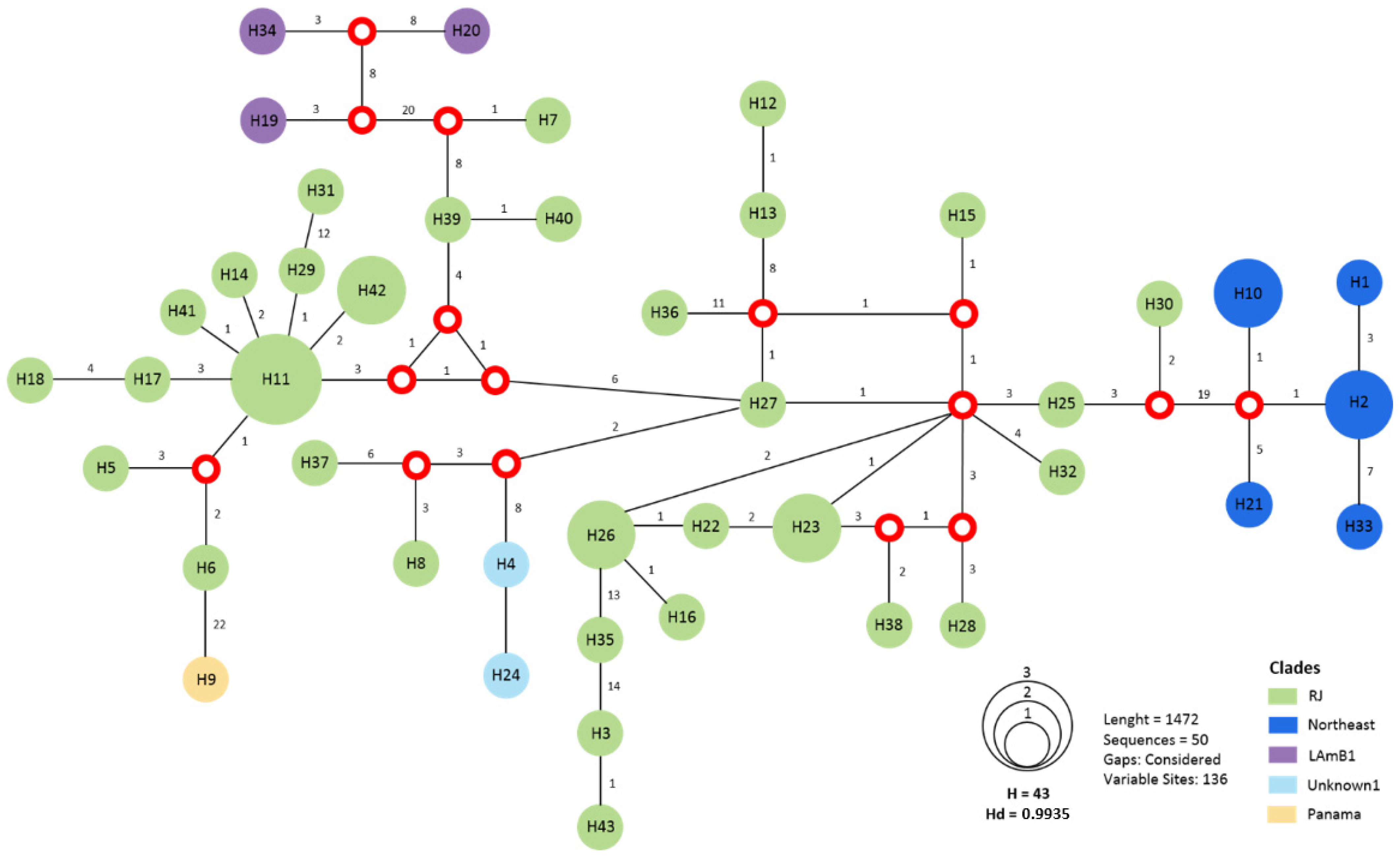
3.3. MLST Is Sufficient to Identify the RJ Clade
3.4. Multiple Infections Caused by Histoplasma spp.
4. Discussion
4.1. Histoplasma Genotypes from Southeast Brazil, Rio de Janeiro
4.2. Strong Population Structure in H. suramericanum in South America
4.3. South America Harbors at Least Two Different Species of Histoplasma
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miranda, C.; Jaker, M.A.; Fitzhugh-Kull, V.A.; Dever, L.L. Oropharyngeal histoplasmosis: The diagnosis lies in the biopsy. IDCases 2018, 11, 33–35. [Google Scholar] [CrossRef]
- Adenis, A.A.; Valdes, A.; Cropet, C.; McCotter, O.Z.; Derado, G.; Couppie, P.; Chiller, T.; Nacher, M. Burden of HIV-associated histoplasmosis compared with tuberculosis in Latin America: A modelling study. Lancet Infect. Dis. 2018, 18, 1150–1159. [Google Scholar] [CrossRef]
- Deepe, G.S., Jr. Outbreaks of histoplasmosis: The spores set sail. PLoS Pathog 2018, 14, e1007213. [Google Scholar] [CrossRef] [PubMed]
- Hage, C.A.; Knox, K.S.; Wheat, L.J. Endemic mycoses: Overlooked causes of community acquired pneumonia. Respir. Med. 2012, 106, 769–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, M.A.; Almeida-Silva, F.; Guimaraes, A.J.; Almeida-Paes, R.; Zancope-Oliveira, R.M. The occurrence of histoplasmosis in Brazil: A systematic review. Int. J. Infect. Dis. 2019, 86, 47–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheat, L.J.; Azar, M.M.; Bahr, N.C.; Spec, A.; Relich, R.F.; Hage, C. Histoplasmosis. Infect. Dis Clin. North. Am. 2016, 30, 207–227. [Google Scholar] [CrossRef]
- Faiolla, R.C.; Coelho, M.C.; Santana Rde, C.; Martinez, R. Histoplasmosis in immunocompetent individuals living in an endemic area in the Brazilian Southeast. Rev. Soc. Bras. Med. Trop. 2013, 46, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, E. Histoplasmose. Mem Inst. Oswaldo Cruz 1945, 43, 457–494. [Google Scholar] [CrossRef] [Green Version]
- Aidé, M.A. Chapter 4–Histoplasmosis. J Bras Pneumol. 2009, 35, 1145–1151. [Google Scholar] [CrossRef] [Green Version]
- Fava, S.D.C.; Fava Netto, C. Epidemiologic Surveys of Histoplasmin and Paracoccidioidin Sensitivity in Brazil. Rev. Inst. Med. Trop. S. Paulo 1998, 40, 155–164. [Google Scholar] [CrossRef]
- Guerra, B.T.; Almeida-Silva, F.; Almeida-Paes, R.; Basso, R.P.; Bernardes, J.P.R.A.; Almeida, M.A.; Damasceno, L.S.; Xavier, M.O.; Wanke, B.; Zancopé-Oliveira, R.M.; et al. Histoplasmosis in Brazil: Lessons to learn about preventing exposure. Mycopathologia 2020, 185, 881–892. [Google Scholar] [CrossRef]
- Almeida, M.A.; Damasceno, L.S.; Pizzini, C.V.; Muniz, M.M.; Almeida-Paes, R.; Zancope-Oliveira, R.M. Role of western blot assay for the diagnosis of histoplasmosis in AIDS patients from a National Institute of Infectious Diseases in Rio de Janeiro, Brazil. Mycoses 2019, 62, 261–267. [Google Scholar] [CrossRef]
- Leimann, B.C.; Pizzini, C.V.; Muniz, M.M.; Albuquerque, P.C.; Monteiro, P.C.; Reis, R.S.; Almeida-Paes, R.; Lazera, M.S.; Wanke, B.; Perez, M.A.; et al. Histoplasmosis in a Brazilian center: Clinical forms and laboratory tests. Rev. Iberoam Micol. 2005, 22, 141–146. [Google Scholar] [CrossRef]
- Kasuga, T.; White, T.J.; Koenig, G.; McEwen, J.; Restrepo, A.; Castaneda, E.; Da Silva Lacaz, C.; Heins-Vaccari, E.M.; De Freitas, R.S.; Zancope-Oliveira, R.M.; et al. Phylogeography of the fungal pathogen Histoplasma capsulatum. Mol. Ecol. 2003, 12, 3383–3401. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda, V.E.; Marquez, R.; Turissini, D.A.; Goldman, W.E.; Matute, D.R. Genome Sequences Reveal Cryptic Speciation in the Human Pathogen Histoplasma capsulatum. MBio 2017, 8, e01339-17. [Google Scholar] [CrossRef] [Green Version]
- Teixeira Mde, M.; Patane, J.S.; Taylor, M.L.; Gomez, B.L.; Theodoro, R.C.; de Hoog, S.; Engelthaler, D.M.; Zancope-Oliveira, R.M.; Felipe, M.S.; Barker, B.M. Worldwide Phylogenetic Distributions and Population Dynamics of the Genus Histoplasma. PLoS Negl. Trop. Dis. 2016, 10, e0004732. [Google Scholar] [CrossRef] [Green Version]
- Damasceno, L.S.; Teixeira, M.D.M.; Barker, B.M.; Almeida, M.A.; Muniz, M.D.M.; Pizzini, C.V.; Mesquita, J.R.L.; Rodríguez-Arellanes, G.; Ramírez, J.A.; Vite-Garín, T.; et al. Novel clinical and dual infection by Histoplasma capsulatum genotypes in HIV patients from Northeastern, Brazil. Sci. Rep. 2019, 9, 11789. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 23 May 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahl, J.W.; Lemmer, D.; Travis, J.; Schupp, J.M.; Gillece, J.D.; Aziz, M.; Driebe, E.M.; Drees, K.P.; Hicks, N.D.; Williamson, C.H.D.; et al. NASP: An accurate, rapid method for the identification of SNPs in WGS datasets that supports flexible input and output formats. Microb. Genom. 2016, 2, e000074. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Nguyen, M.A.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2013. [Google Scholar]
- Champely, S.; Ekstrom, C.; Dalgaard, P.; Gill, J.; Weibelzahl, S.; Anandkumar, A.; Ford, C.; Volcic, R.; De Rosario, H. Basic Functions for Power Analysis, Package ‘pwr’. 2020. Available online: https://cran.r-project.org/web/packages/pwr/ (accessed on 25 July 2021).
- Munoz, M.; Wintaco, L.M.; Munoz, S.A.; Ramirez, J.D. Dissecting the Heterogeneous Population Genetic Structure of Candida albicans: Limitations and Constraints of the Multilocus Sequence Typing Scheme. Front. Microbiol. 2019, 10, 1052. [Google Scholar] [CrossRef]
- Vincent, R.D.; Goewert, R.; Goldman, W.E.; Kobayashi, G.S.; Lambowitz, A.M.; Medoff, G. Classification of Histoplasma capsulatum isolates by restriction fragment polymorphisms. J. Bacteriol. 1986, 165, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Kasuga, T.; Taylor, J.W.; White, T.J. Phylogenetic relationships of varieties and geographical groups of the human pathogenic fungus Histoplasma capsulatum Darling. J. Clin. Microbiol. 1999, 37, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Ropars, J.; Maufrais, C.; Diogo, D.; Marcet-Houben, M.; Perin, A.; Sertour, N.; Mosca, K.; Permal, E.; Laval, G.; Bouchier, C.; et al. Gene flow contributes to diversification of the major fungal pathogen Candida albicans. Nat. Commun. 2018, 9, 2253. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, C.S.; Sepulveda, V.E.; Turissini, D.A.; Goldman, W.E.; Matute, D.R. Recent admixture between species of the fungal pathogen Histoplasma. Evol. Lett. 2018, 2, 210–220. [Google Scholar] [CrossRef]
- Sepulveda, V.E.; Williams, C.L.; Goldman, W.E. Comparison of phylogenetically distinct Histoplasma strains reveals evolutionarily divergent virulence strategies. MBio 2014, 5, e01376-01314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.S.; Sepulveda, V.E.; Goldman, W.E. Biodiverse Histoplasma Species Elicit Distinct Patterns of Pulmonary Inflammation following Sublethal Infection. mSphere 2020, 5, e00742-20. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.G.; Silva, R.S.; Carniello, M.A.; Veldman, J.W.; Rossi, A.A.; de Oliveira, L.O. Molecular evidence of cryptic speciation, historical range expansion, and recent intraspecific hybridization in the Neotropical seasonal forest tree Cedrela fissilis (Meliaceae). Mol. Phylogenet Evol. 2011, 61, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batalha-Filho, H.; Maldonado-Coelho, M.; Miyaki, C.Y. Historical climate changes and hybridization shaped the evolution of Atlantic Forest spinetails (Aves: Furnariidae). Heredity 2019, 123, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves-Oliveira, J.; Teixeira, B.R.; Olifiers, N.; dos Santos Lucio, C.; Riski, L.L.; da Costa-Neto, S.F.; de Lemos, E.R.S.; Bonvicino, C.R.; D’Andrea, P.S. A Survey of Small Mammals in the Atlantic Forest Of the Northwestern Region Of Rio De Janeiro State. Oecologia Aust. 2016, 20, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Matute, D.R.; Sepulveda, V.E. Fungal species boundaries in the genomics era. Fungal Genet. Biol. 2019, 131, 103249. [Google Scholar] [CrossRef]
- Gomez, L.F.; Arango, M.; McEwen, J.G.; Gomez, O.M.; Zuluaga, A.; Pelaez, C.A.; Acevedo, J.M.; Taylor, M.L.; Jimenez, M.D.P. Molecular epidemiology of Colombian Histoplasma capsulatum isolates obtained from human and chicken manure samples. Heliyon 2019, 5, e02084. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Patient | Isolate | Origin | Specimen | Clinical Features | Phylogenetic Species |
|---|---|---|---|---|---|
| 1 | 13H | Human | Blood Culture | HIV+/Disseminated | NE |
| 2 | 129H | Human | Blood Culture | HIV+/Disseminated | NE |
| 3 | 20231 | Human | Tongue Scraping | HIV+/Disseminated | Unknown1 |
| - | 11354 | Human | Blood Culture | HIV+/Disseminated | Unknown1 |
| 4 | 3416 | Human | Sputum | HIV−/Chronic Pulmonary | RJ |
| - | 3612 | Human | Lung Biopsy | HIV+/Disseminated | RJ |
| - | 39439 | Human | Bone Marrow Aspirate | HIV+/Disseminated | RJ |
| - | 39942 | Human | Biopsy | NA | Panama |
| 5 | 6503 | Human | Ganglion Biopsy | HIV+/Opportunistic | RJ |
| - | 84502 | Human | Blood Culture | HIV+/Disseminated | NE |
| - | 84564 | Human | Skin Biopsy | HIV+/Disseminated | NE |
| - | AC 05 | Environmental | Soil | NA | RJ |
| - | CÃO 4 | Animal/Dog | Liver and Spleen | NA | RJ |
| - | CO4 | Environmental | Soil | NA | RJ |
| - | EP 02 | Environmental | Soil | NA | RJ |
| 1 | HC 18 | Human | Blood Culture | HIV+/Disseminated | RJ |
| 6 | 40039 | Human | Bone Marrow Aspirate | HIV+/Disseminated | RJ |
| - | IGS 19 | Environmental | Soil | NA | RJ |
| - | IGS 4/5 | Environmental | Soil | NA | RJ |
| 7 | INI 01/16 | Human | Bone Marrow Aspirate | HIV+/Disseminated | NE |
| 8 | INI 02/16 | Human | Skin Biopsy | HIV+/Disseminated | LAmB1 |
| 8 | INI 03/16 | Human | Bone Marrow Aspirate | HIV+/Disseminated | LAmB1 |
| 9 | INI 04/16 | Human | Sputum | HIV-/Disseminated | NE |
| 9 | INI 05/16 | Human | Ganglion Fragment | HIV-/Disseminated | RJ |
| 10 | INI 06/16 | Human | Bone Marrow Aspirate | HIV+/Disseminated | RJ |
| 11 | INI 07/16 | Human | Bone Marrow Aspirate | HIV+/Disseminated | Unknown1 |
| 12 | 01_12 | Human | Bone Marrow Aspirate | HIV+/Disseminated | RJ |
| 13 | 01_13 | Human | Blood Culture | HIV+/Disseminated | RJ |
| - | 02_13 | Animal/Dog | Blood Culture | NA | RJ |
| 14 | 02_14 | Human | Skin Biopsy | HIV+/Disseminated | RJ |
| 15 | 04_12 | Human | Bone Marrow Aspirate | HIV+/Disseminated | RJ |
| 16 | 04_14 | Human | Bone Marrow Aspirate | HIV+/Disseminated | RJ |
| - | 05_12 | Animal/Cat | Lesion Exudate | NA | RJ |
| 17 | 06_12 | Human | Bone Marrow Aspirate | HIV+/Disseminated | RJ |
| - | 07_12 | Animal/Dog | Lymph Node Biopsy | NA | RJ |
| 18 | 09_12 | Human | Bronchoalveolar Lavage | Chronic Pulmonary | NE |
| 19 | 11_12 | Human | Skin Biopsy | HIV+/Disseminated | LAmB1 |
| 13 | 23_11 | Human | Blood Culture | HIV+/Disseminated | Unknown1 |
| - | 24_11 | Human | Blood Culture | HIV+/Disseminated | RJ |
| - | 26_11 | Human | Bone Marrow Aspirate | HIV+/Disseminated | RJ |
| 20 | 27_11 | Human | Oropharyngeal Swab | HIV+/Disseminated | RJ |
| 20 | 28_11 | Human | Bone Marrow Aspirate | HIV+/Disseminated | RJ |
| - | IT 04 | Environmental | Soil | NA | RJ |
| - | RPS 35 | Environmental | Soil | NA | RJ |
| - | RPS 45 | Environmental | Soil | NA | RJ |
| - | RPS 51 | Environmental | Soil | NA | RJ |
| - | RS 36 | Wild Animal | NA | NA | RJ |
| - | TI 01 | Environmental | Soil | NA | RJ |
| - | TI 05 | Environmental | Soil | NA | RJ |
| - | 36GAL | Human | Blood | HIV+/Disseminated | Unknown1 |
| Clinical Data | Genotype | Power | p-Value | Corrected p-Value | |
|---|---|---|---|---|---|
| RJ | Others | ||||
| Fever | 9 (90%) | 5 (71.4%) | 0.166 | ||
| Weight Loss | 6 (60%) | 5 (71.4%) | 0.078 | ||
| Cough | 6 (60%) | 6 (85.7%) | 0.226 | ||
| Dyspnea | 3 (30%) | 1 (14.2%) | 0.122 | ||
| Abdominal Pain | 3 (30%) | 2 (28.5%) | 0.050 | ||
| Diarrhea | 1 (10%) | 4 (57.1%) | 0.584 | 0.119 | 0.476 |
| Vomit | 3 (30%) | 3 (42.8%) | 0.085 | ||
| Asthenia | 3 (30%) | 5 (71.4%) | 0.411 | 0.234 | 0.701 |
| Headache | 0 (0%) | 2 (28.5%) | 0.629 | 0.301 | 0.701 |
| Hepatomegaly | 4 (40%) | 2 (28.5%) | 0.078 | ||
| Splenomegaly | 5 (50%) | 4 (57.1%) | 0.060 | ||
| Acute Renal Failure | 7 (70%) | 2 (28.5%) | 0.411 | 0.234 | 0.701 |
| Hemorrhage | 3 (30%) | 1 (14.2%) | 0.122 | ||
| Skin Lesion | 4 (40%) | 3 (42.8%) | 0.0516 | ||
| Adenomegaly | 6 (60%) | 4 (57.1%) | 0.0516 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida-Silva, F.; de Melo Teixeira, M.; Matute, D.R.; de Faria Ferreira, M.; Barker, B.M.; Almeida-Paes, R.; Guimarães, A.J.; Zancopé-Oliveira, R.M. Genomic Diversity Analysis Reveals a Strong Population Structure in Histoplasma capsulatum LAmA (Histoplasma suramericanum). J. Fungi 2021, 7, 865. https://doi.org/10.3390/jof7100865
Almeida-Silva F, de Melo Teixeira M, Matute DR, de Faria Ferreira M, Barker BM, Almeida-Paes R, Guimarães AJ, Zancopé-Oliveira RM. Genomic Diversity Analysis Reveals a Strong Population Structure in Histoplasma capsulatum LAmA (Histoplasma suramericanum). Journal of Fungi. 2021; 7(10):865. https://doi.org/10.3390/jof7100865
Chicago/Turabian StyleAlmeida-Silva, Fernando, Marcus de Melo Teixeira, Daniel R. Matute, Marcela de Faria Ferreira, Bridget M. Barker, Rodrigo Almeida-Paes, Allan J. Guimarães, and Rosely M. Zancopé-Oliveira. 2021. "Genomic Diversity Analysis Reveals a Strong Population Structure in Histoplasma capsulatum LAmA (Histoplasma suramericanum)" Journal of Fungi 7, no. 10: 865. https://doi.org/10.3390/jof7100865