
Genome Comparisons of the Fission Yeasts Reveal Ancient Collinear Loci Maintained by Natural Selection

Abstract
:1. Introduction
2. Materials and Methods
2.1. Species and Genomes Data
2.2. Whole Genome Alignments and Sequence Comparisons
2.3. Genome Rearrangement Analyses
2.4. Orthology Inference
2.5. Visualization of Collinear Blocks
2.6. Phylogenetic Tree Constructions
2.7. Modelling Genome Evolution
2.8. Study of Evolutionary Rates and Intron Loss/Gain
2.9. GO Enrichment Analyses
2.10. Density of Essential Genes and Rec12 Cleavage Sites
2.11. Analyses of Co-Expression Rates
2.12. Normalisation and Randomisation
2.13. Statistical Analyses
2.14. Image Creation
3. Results
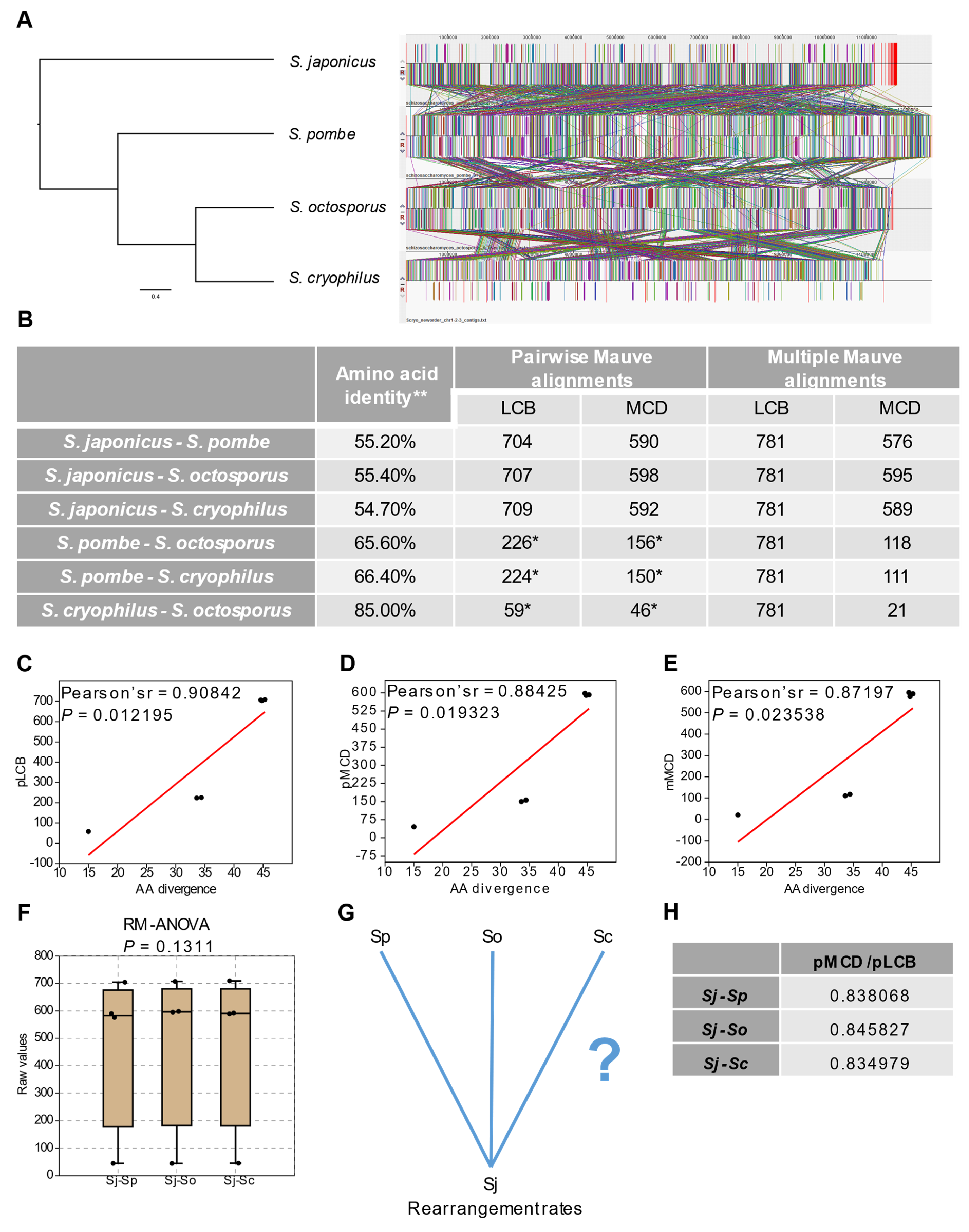
3.1. Structural- and Sequence Evolution of the Fission Yeasts Show Unequivocal Correlations despite Their Evolutionary Breadth
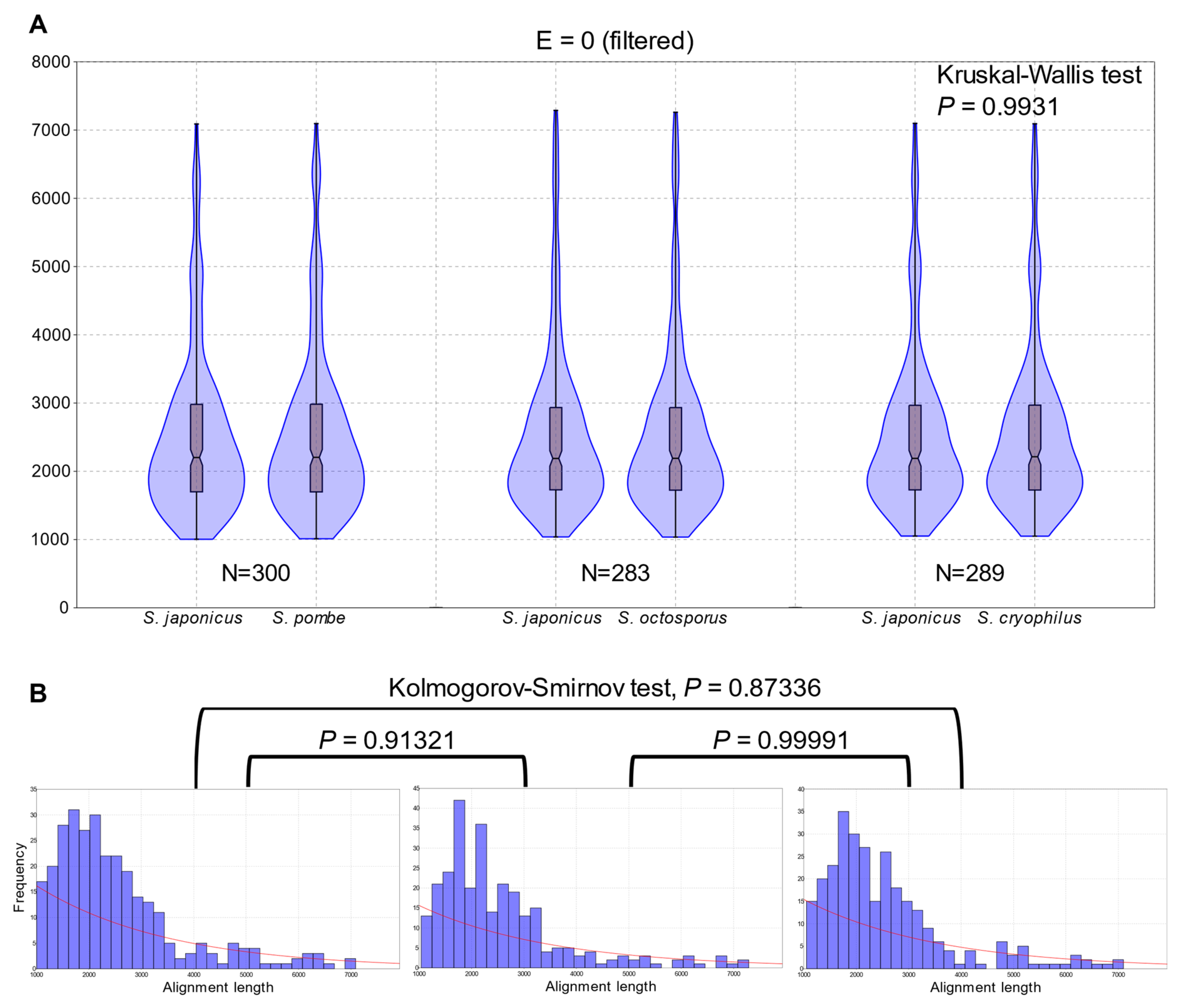
3.2. Extent of Whole Genome Conservation of the Fission Yeasts Is Almost Equal
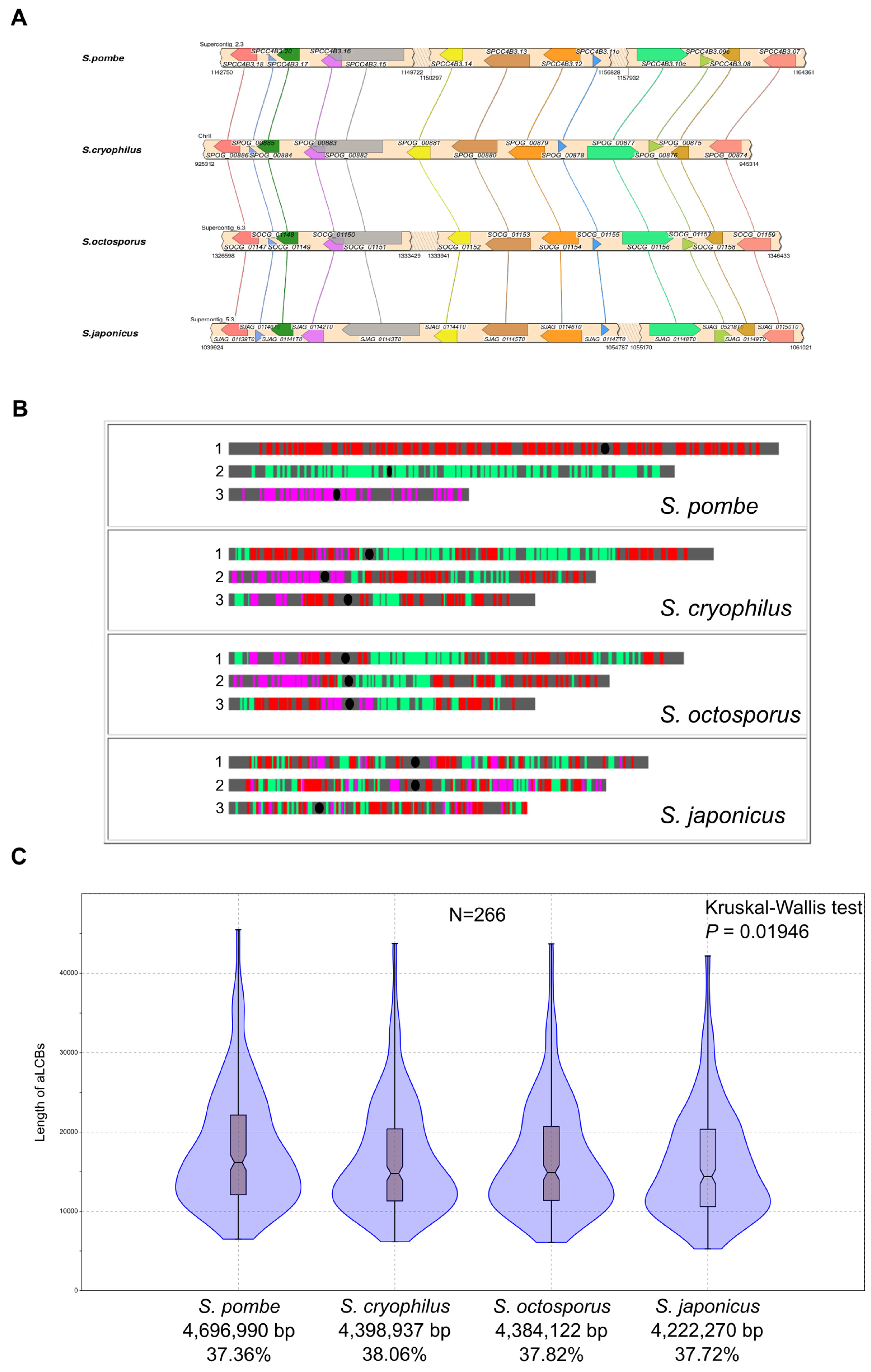
3.3. Analyses of Gene Level Genome Conservation Reveals Ancient Collinear Loci Inherited from the Last Common Ancestor of the Fission Yeasts
3.4. The aLCBs Are Not Only Remnants of Ancestral Gene Order but Are Also Maintained by Natural Selection
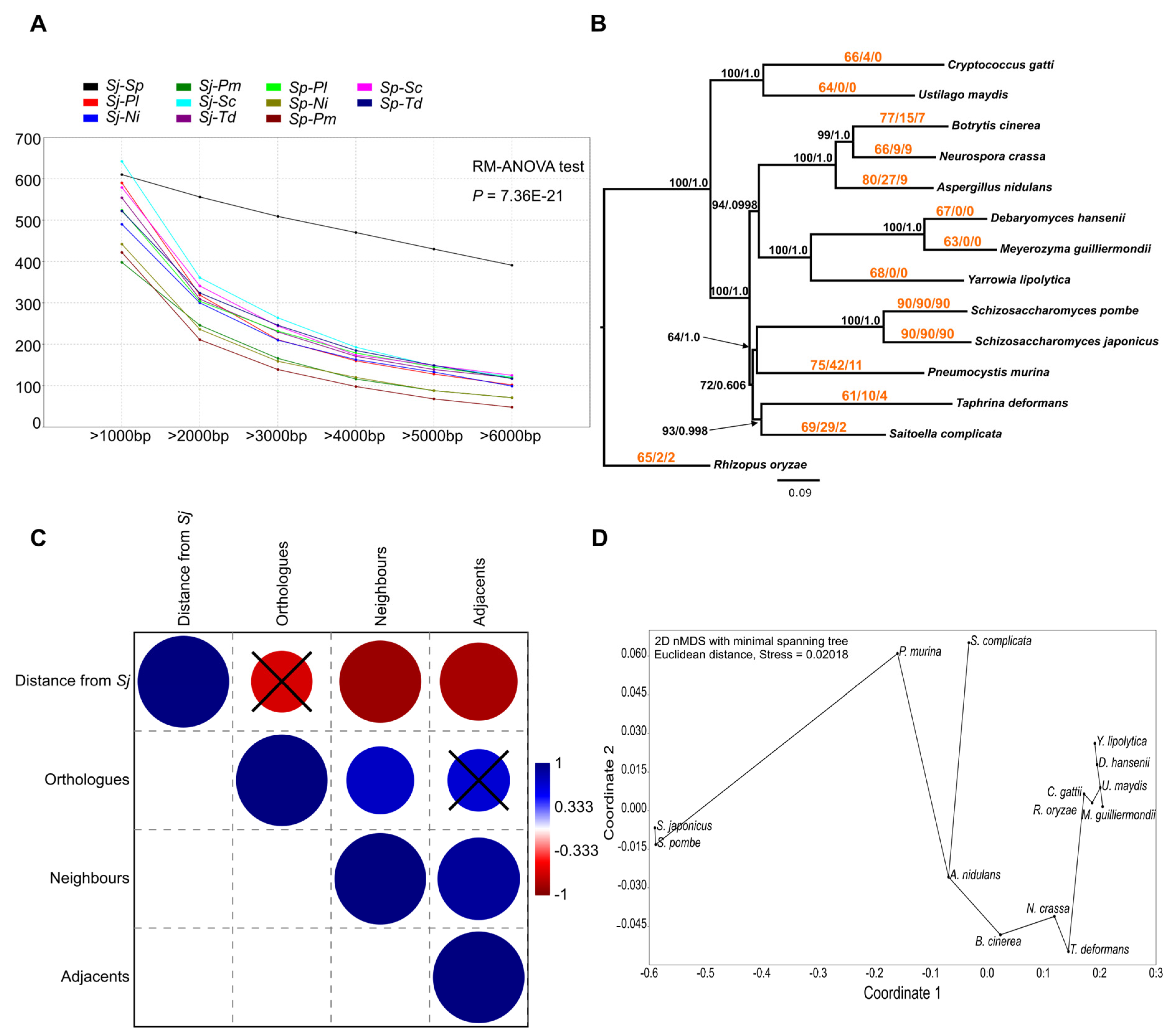
3.5. Gene Order of the aLCBs Is Mainly Fission-Yeast-Specific but Further Supports the Idea of Filamentous Ancestors
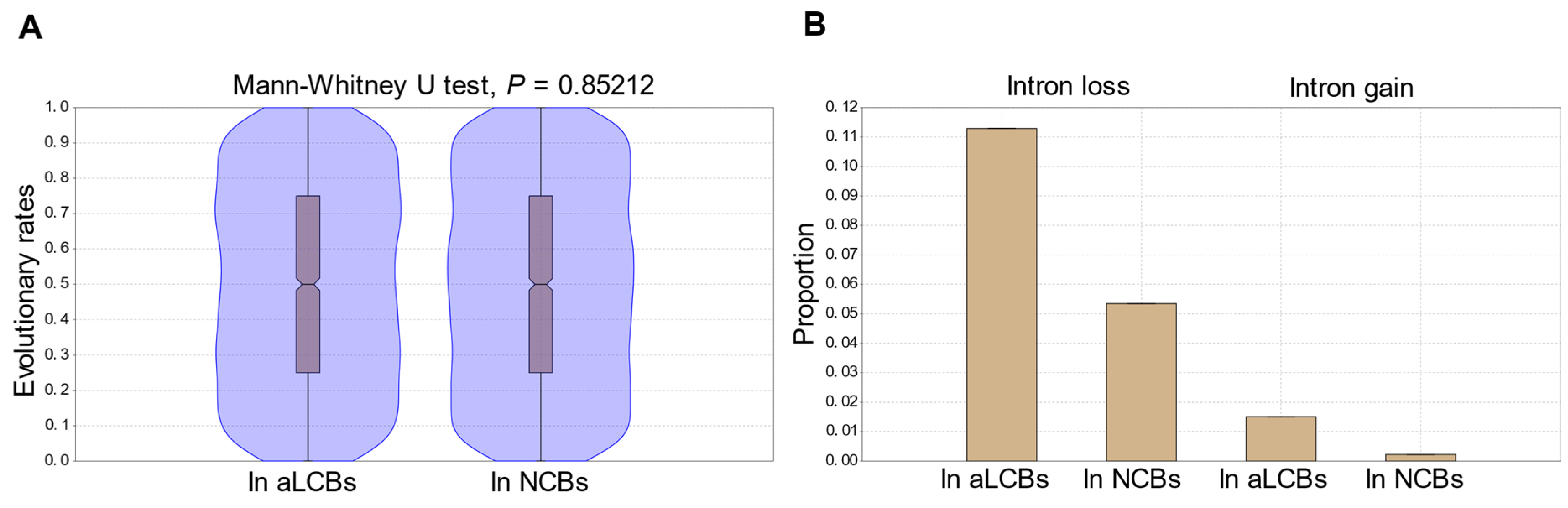
3.6. Comparisons of Sequence- and Gene Structural Changes Indicate That Gene Sequences in the aLCBs Are as Mutable as Any Sequences in Other Parts of the Genomes
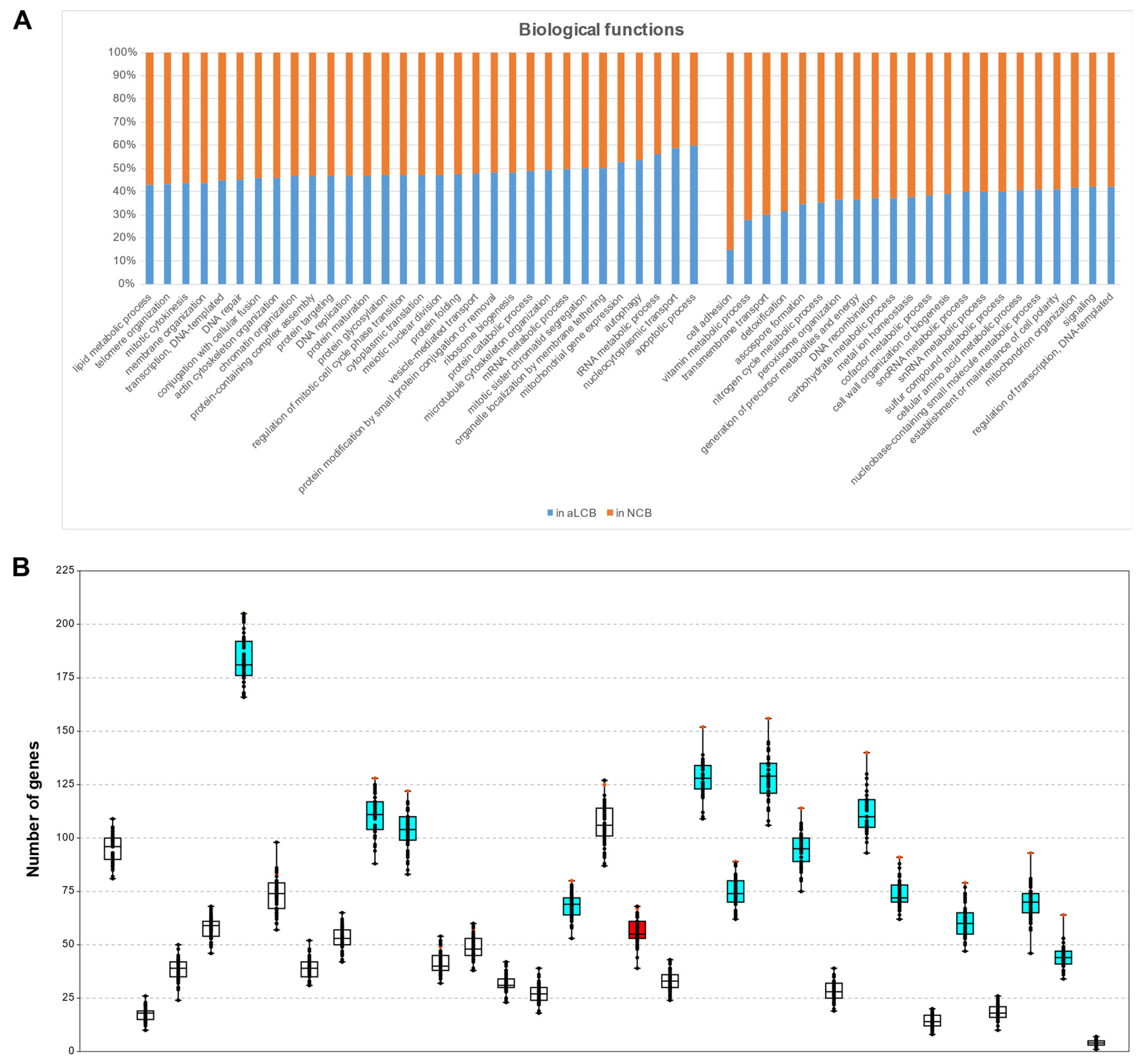
3.7. Genes of Certain GO Categories Tend to Cluster to the aLCBs
3.8. Co-Expression Rates of the Genes in the aLCBs Are Not Higher Than the Co-Expression Rates of Other Genes Outside the aLCBs
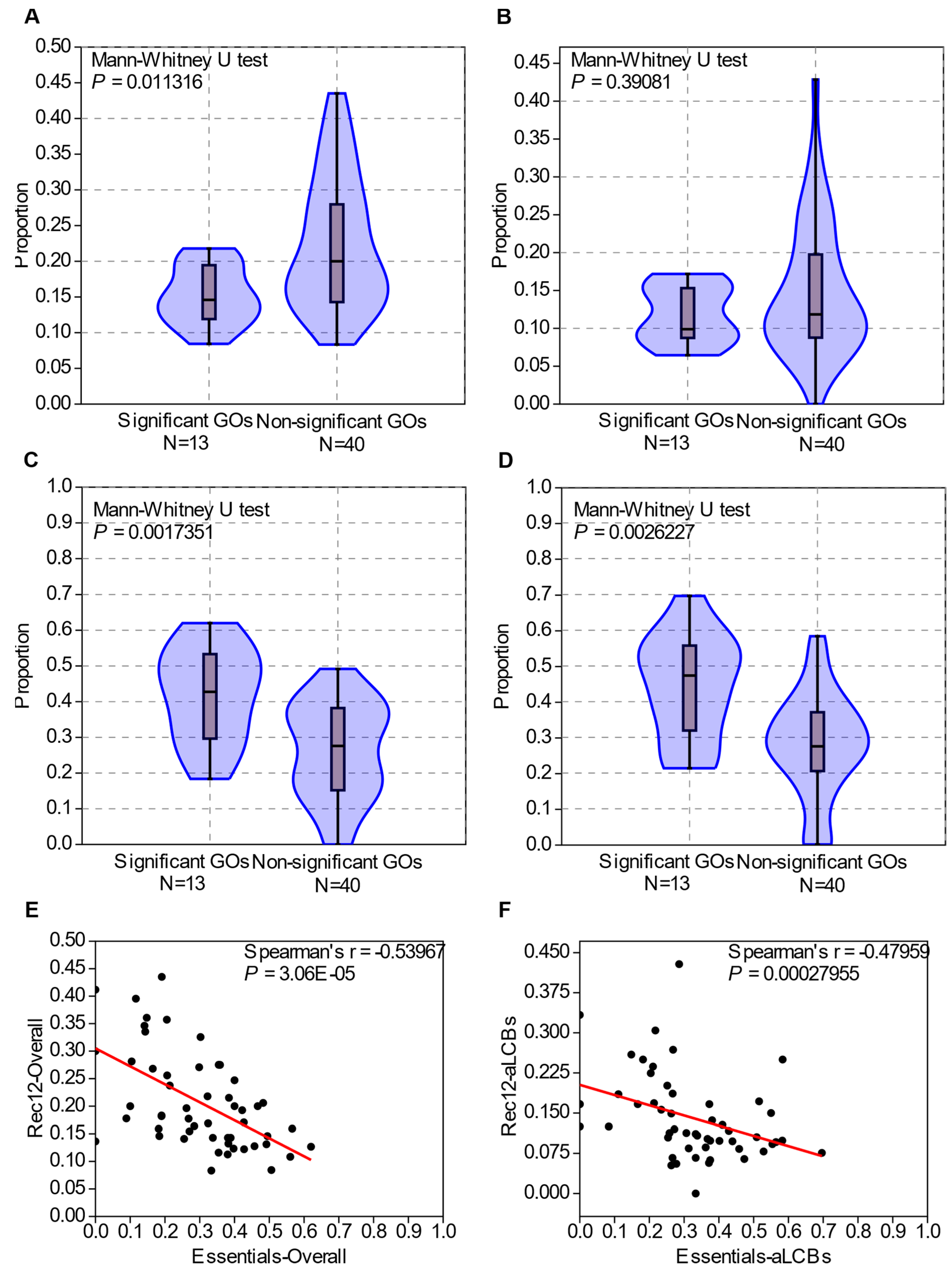
3.9. GO Enrichment in the aLCBs Is Not Related to the Biological Functions, It Is Rather Determined by the Density of Essential Genes and Rec12 Cleavage Sites
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Y.; Leigh, J.W.; Brinkmann, H.; Cushion, M.T.; Rodriguez-Ezpeleta, N.; Philippe, H.; Lang, B.F. Phylogenomic analyses support the monophyly of Taphrinomycotina, including Schizosaccharomyces fission yeasts. Mol. Biol. Evol. 2009, 26, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Rhind, N.; Chen, Z.; Yassour, M.; Thompson, D.A.; Haas, B.J.; Habib, N.; Wapinski, I.; Roy, S.; Lin, M.F.; Heiman, D.I.; et al. Comparative functional genomics of the fission yeasts. Science 2011, 332, 930–936. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.X.; Steenwyk, J.L.; LaBella, A.L.; Opulente, D.A.; Zhou, X.; Kominek, J.; Li, Y.; Groenewald, M.; Hittinger, C.T.; Rokas, A. Genome-scale phylogeny and contrasting modes of genome evolution in the fungal phylum Ascomycota. Sci. Adv. 2020, 6, eabd0079. [Google Scholar] [CrossRef]
- Li, Y.; Steenwyk, J.L.; Chang, Y.; Wang, Y.; James, T.Y.; Stajich, J.E.; Spatafora, J.W.; Groenewald, M.; Dunn, C.W.; Hittinger, C.T.; et al. A genome-scale phylogeny of the kingdom Fungi. Curr. Biol. 2021, 31, 1653–1665.e5. [Google Scholar] [CrossRef]
- Sipiczki, M. Where does fission yeast sit on the tree of life? Genome Biol. 2000, 1, reviews1011.1. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z. Big data mining powers fungal research: Recent advances in fission yeast systems biology approaches. Curr. Genet. 2017, 63, 427–433. [Google Scholar] [CrossRef]
- Hayles, J.; Nurse, P. Introduction to Fission Yeast as a Model System. Cold Spring Harb. Protoc. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, C.S.; Wood, V.; Fantes, P.A. An Ancient Yeast for Young Geneticists: A Primer on the Schizosaccharomyces pombe Model System. Genetics 2015, 201, 403–423. [Google Scholar] [CrossRef] [Green Version]
- Brown, W.R.; Liti, G.; Rosa, C.; James, S.; Roberts, I.; Robert, V.; Jolly, N.; Tang, W.; Baumann, P.; Green, C.; et al. A Geographically Diverse Collection of Schizosaccharomyces pombe Isolates Shows Limited Phenotypic Variation but Extensive Karyotypic Diversity. G3 Genes Genomes Genet. 2011, 1, 615–626. [Google Scholar]
- Avelar, A.T.; Perfeito, L.; Gordo, I.; Ferreira, M.G. Genome architecture is a selectable trait that can be maintained by antagonistic pleiotropy. Nat. Commun. 2013, 4, 2235. [Google Scholar] [CrossRef] [Green Version]
- Fawcett, J.A.; Iida, T.; Takuno, S.; Sugino, R.P.; Kado, T.; Kugou, K.; Mura, S.; Kobayashi, T.; Ohta, K.; Nakayama, J.; et al. Population genomics of the fission yeast Schizosaccharomyces pombe. PLoS ONE 2014, 9, e104241. [Google Scholar] [CrossRef] [Green Version]
- Zanders, S.E.; Eickbush, M.T.; Yu, J.S.; Kang, J.W.; Fowler, K.R.; Smith, G.R.; Malik, H.S. Genome rearrangements and pervasive meiotic drive cause hybrid infertility in fission yeast. eLife 2014, 3, e02630. [Google Scholar] [CrossRef]
- Naumov, G.I.; Kondratieva, V.I.; Naumova, E.S. Hybrid Sterility of the Yeast Schizosaccharomyces pombe: Genetic Genus and Many Species in statu nascendi? Mikrobiologiia 2015, 84, 192–203. [Google Scholar] [CrossRef]
- Jeffares, D.C.; Rallis, C.; Rieux, A.; Speed, D.; Převorovský, M.; Mourier, T.; Marsellach, F.X.; Iqbal, Z.; Lau, W.; Cheng, T.M.; et al. The genomic and phenotypic diversity of Schizosaccharomyces pombe. Nat. Genet. 2015, 47, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Jeffares, D.C.; Jolly, C.; Hoti, M.; Speed, D.; Shaw, L.; Rallis, C.; Balloux, F.; Dessimoz, C.; Bähler, J.; Sedlazeck, F.J. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 2017, 8, 14061. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.T.; Suo, F.; Tusso, S.; Wang, Y.K.; Huang, S.; Wolf, J.B.W.; Du, L.L. Intraspecific Diversity of Fission Yeast Mitochondrial Genomes. Genome Biol. Evol. 2019, 11, 2312–2329. [Google Scholar] [CrossRef] [Green Version]
- Tusso, S.; Nieuwenhuis, B.P.S.; Sedlazeck, F.J.; Davey, J.W.; Jeffares, D.C.; Wolf, J.B.W. Ancestral Admixture Is the Main Determinant of Global Biodiversity in Fission Yeast. Mol. Biol. Evol. 2019, 36, 1975–1989. [Google Scholar] [CrossRef] [Green Version]
- Sipiczki, M.; Takeo, K.; Yamaguchi, M.; Yoshida, S.; Miklos, I. Environmentally controlled dimorphic cycle in a fission yeast. Microbiology 1998, 144 Pt 5, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Balazs, A.; Batta, G.; Miklos, I.; Acs-Szabo, L.; Vazquez de Aldana, C.R.; Sipiczki, M. Conserved regulators of the cell separation process in Schizosaccharomyces. Fungal Genet. Biol. 2012, 49, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Klar, A.J. Schizosaccharomyces japonicus yeast poised to become a favorite experimental organism for eukaryotic research. G3 Genes Genomes Genet. 2013, 3, 1869–1873. [Google Scholar]
- Niki, H. Schizosaccharomyces japonicus: The fission yeast is a fusion of yeast and hyphae. Yeast 2014, 31, 83–90. [Google Scholar] [CrossRef]
- Makarova, M.; Gu, Y.; Chen, J.S.; Beckley, J.R.; Gould, K.L.; Oliferenko, S. Temporal Regulation of Lipin Activity Diverged to Account for Differences in Mitotic Programs. Curr. Biol. 2016, 26, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Gil, E.; Franco, A.; Vázquez-Marín, B.; Prieto-Ruiz, F.; Pérez-Díaz, A.; Vicente-Soler, J.; Madrid, M.; Soto, T.; Cansado, J. Specific Functional Features of the Cell Integrity MAP Kinase Pathway in the Dimorphic Fission Yeast Schizosaccharomyces japonicus. J. Fungi 2021, 7, 482. [Google Scholar] [CrossRef]
- Seike, T.; Sakata, N.; Matsuda, F.; Furusawa, C. Elevated Sporulation Efficiency in Fission Yeast Schizosaccharomyces japonicus Strains Isolated from Drosophila. J. Fungi 2021, 7, 350. [Google Scholar] [CrossRef]
- Papp, L.A.; Ács-Szabó, L.; Batta, G.; Miklós, I. Molecular and comparative genomic analyses reveal evolutionarily conserved and unique features of the Schizosaccharomyces japonicus mycelial growth and the underlying genomic changes. Curr. Genet. 2021. [Google Scholar] [CrossRef]
- Harmouch, N.; Coulon, J.; Bonaly, R. Identification of 24-methylene-24,25-dihydrolanosterol as a precursor of ergosterol in the yeasts Schizosaccharomyces pombe and Schizosaccharomyces octosporus. FEMS Microbiol. Lett. 1995, 134, 147–152. [Google Scholar] [CrossRef]
- Bullerwell, C.E.; Leigh, J.; Forget, L.; Lang, B.F. A comparison of three fission yeast mitochondrial genomes. Nucleic Acids Res. 2003, 31, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Seike, T.; Niki, H. Mating response and construction of heterothallic strains of the fission yeast Schizosaccharomyces octosporus. FEMS Yeast Res. 2017, 17, fox045. [Google Scholar] [CrossRef]
- Helston, R.M.; Box, J.A.; Tang, W.; Baumann, P. Schizosaccharomyces cryophilus sp. nov., a new species of fission yeast. FEMS Yeast Res. 2010, 10, 779–786. [Google Scholar] [CrossRef] [Green Version]
- Ács-Szabó, L.; Papp, L.A.; Antunovics, Z.; Sipiczki, M.; Miklós, I. Assembly of Schizosaccharomyces cryophilus chromosomes and their comparative genomic analyses revealed principles of genome evolution of the haploid fission yeasts. Sci. Rep. 2018, 8, 14629. [Google Scholar] [CrossRef]
- Brysch-Herzberg, M.; Tobias, A.; Seidel, M.; Wittmann, R.; Wohlmann, E.; Fischer, R.; Dlauchy, D.; Peter, G. Schizosaccharomyces osmophilus sp. nov., an osmophilic fission yeast occurring in bee bread of different solitary bee species. FEMS Yeast Res. 2019, 19, foz038. [Google Scholar] [CrossRef]
- Kudriawzew, W. Die Systematik der Hefen; Akademie: Berlin, Germany, 1960. [Google Scholar]
- Yamada, Y.; Banno, I. Hasegawaea gen. nov., an ascosporoge-nous yeast genus for the organisms whose asexual reproduction is by fission and whose ascospores have smooth surfaces without papillae and which are characterized by the absence of coenzyme Q mid by the presence of linoleic acid in cellular fatty acid composition. J. Gen. Appl. Microbiol. 1987, 33, 295–298. [Google Scholar]
- Wood, V.; Gwilliam, R.; Rajandream, M.A.; Lyne, M.; Lyne, R.; Stewart, A.; Sgouros, J.; Peat, N.; Hayles, J.; Baker, S.; et al. The genome sequence of Schizosaccharomyces pombe. Nature 2002, 415, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Tong, P.; Pidoux, A.L.; Toda, N.R.T.; Ard, R.; Berger, H.; Shukla, M.; Torres-Garcia, J.; Müller, C.A.; Nieduszynski, C.A.; Allshire, R.C. Interspecies conservation of organisation and function between nonhomologous regional centromeres. Nat. Commun. 2019, 10, 2343. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [Green Version]
- Cissé, O.H.; Almeida, J.M.; Fonseca, A.; Kumar, A.A.; Salojärvi, J.; Overmyer, K.; Hauser, P.M.; Pagni, M. Genome sequencing of the plant pathogen Taphrina deformans, the causal agent of peach leaf curl. mBio 2013, 4, e00055-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, R.; Haridas, S.; Wolfe, K.H.; Lopes, M.R.; Hittinger, C.T.; Göker, M.; Salamov, A.A.; Wisecaver, J.H.; Long, T.M.; Calvey, C.H.; et al. Comparative genomics of biotechnologically important yeasts. Proc. Natl. Acad. Sci. USA 2016, 113, 9882–9887. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Chen, Z.; da Huang, W.; Kutty, G.; Ishihara, M.; Wang, H.; Abouelleil, A.; Bishop, L.; Davey, E.; Deng, R.; et al. Genome analysis of three Pneumocystis species reveals adaptation mechanisms to life exclusively in mammalian hosts. Nat. Commun. 2016, 7, 10740. [Google Scholar] [CrossRef] [PubMed]
- Mondo, S.J.; Dannebaum, R.O.; Kuo, R.C.; Louie, K.B.; Bewick, A.J.; LaButti, K.; Haridas, S.; Kuo, A.; Salamov, A.; Ahrendt, S.R.; et al. Widespread adenine N6-methylation of active genes in fungi. Nat. Genet. 2017, 49, 964–968. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Cissé, O.H.; Yun Wong, J.; Zheng, P.; Hewitt, D.; Nowrousian, M.; Stajich, J.E.; Jedd, G. Innovation and constraint leading to complex multicellularity in the Ascomycota. Nat. Commun. 2017, 8, 14444. [Google Scholar] [CrossRef] [PubMed]
- Noé, L.; Kucherov, G. YASS: Enhancing the sensitivity of DNA similarity search. Nucleic Acids Res. 2005, 33, W540–W543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesler, G. GRIMM: Genome rearrangements web server. Bioinformatics 2002, 18, 492–493. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- D’Souza, C.A.; Kronstad, J.W.; Taylor, G.; Warren, R.; Yuen, M.; Hu, G.; Jung, W.H.; Sham, A.; Kidd, S.E.; Tangen, K.; et al. Genome variation in Cryptococcus gattii, an emerging pathogen of immunocompetent hosts. mBio 2011, 2, e00342-10. [Google Scholar] [CrossRef] [Green Version]
- Farrer, R.A.; Desjardins, C.A.; Sakthikumar, S.; Gujja, S.; Saif, S.; Zeng, Q.; Chen, Y.; Voelz, K.; Heitman, J.; May, R.C.; et al. Genome Evolution and Innovation across the Four Major Lineages of Cryptococcus gattii. mBio 2015, 6, e00868-15. [Google Scholar] [CrossRef] [Green Version]
- Butler, G.; Rasmussen, M.D.; Lin, M.F.; Santos, M.A.; Sakthikumar, S.; Munro, C.A.; Rheinbay, E.; Grabherr, M.; Forche, A.; Reedy, J.L.; et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 2009, 459, 657–662. [Google Scholar] [CrossRef] [Green Version]
- Galagan, J.E.; Calvo, S.E.; Cuomo, C.; Ma, L.J.; Wortman, J.R.; Batzoglou, S.; Lee, S.I.; Baştürkmen, M.; Spevak, C.C.; Clutterbuck, J.; et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 2005, 438, 1105–1115. [Google Scholar] [CrossRef]
- Galagan, J.E.; Calvo, S.E.; Borkovich, K.A.; Selker, E.U.; Read, N.D.; Jaffe, D.; FitzHugh, W.; Ma, L.J.; Smirnov, S.; Purcell, S.; et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature 2003, 422, 859–868. [Google Scholar] [CrossRef]
- Ma, L.J.; Ibrahim, A.S.; Skory, C.; Grabherr, M.G.; Burger, G.; Butler, M.; Elias, M.; Idnurm, A.; Lang, B.F.; Sone, T.; et al. Genomic analysis of the basal lineage fungus Rhizopus oryzae reveals a whole-genome duplication. PLoS Genet. 2009, 5, e1000549. [Google Scholar] [CrossRef]
- Kämper, J.; Kahmann, R.; Bölker, M.; Ma, L.J.; Brefort, T.; Saville, B.J.; Banuett, F.; Kronstad, J.W.; Gold, S.E.; Müller, O.; et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 2006, 444, 97–101. [Google Scholar] [CrossRef]
- Dujon, B.; Sherman, D.; Fischer, G.; Durrens, P.; Casaregola, S.; Lafontaine, I.; De Montigny, J.; Marck, C.; Neuvéglise, C.; Talla, E.; et al. Genome evolution in yeasts. Nature 2004, 430, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Sacerdot, C.; Casaregola, S.; Lafontaine, I.; Tekaia, F.; Dujon, B.; Ozier-Kalogeropoulos, O. Promiscuous DNA in the nuclear genomes of hemiascomycetous yeasts. FEMS Yeast Res. 2008, 8, 846–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veltri, D.; Wight, M.M.; Crouch, J.A. SimpleSynteny: A web-based tool for visualization of microsynteny across multiple species. Nucleic Acids Res. 2016, 44, W41–W45. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.P.; Vergara, I.A.; Frech, C.; Chen, Q.; Zeng, X.; Pei, J.; Chen, N. OrthoClusterDB: An online platform for synteny blocks. BMC Bioinform. 2009, 10, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, 636–641. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [Green Version]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef]
- Han, M.V.; Zmasek, C.M. phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinform. 2009, 10, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalquen, D.A.; Anisimova, M.; Gonnet, G.H.; Dessimoz, C. ALF—A simulation framework for genome evolution. Mol. Biol. Evol. 2012, 29, 1115–1123. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Niu, D.K. Mechanisms of intron loss and gain in the fission yeast Schizosaccharomyces. PLoS ONE 2013, 8, e61683. [Google Scholar] [CrossRef] [PubMed]
- Lock, A.; Rutherford, K.; Harris, M.A.; Hayles, J.; Oliver, S.G.; Bähler, J.; Wood, V. PomBase 2018: User-driven reimplementation of the fission yeast database provides rapid and intuitive access to diverse, interconnected information. Nucleic Acids Res. 2019, 47, D821–D827. [Google Scholar] [CrossRef] [Green Version]
- Fowler, K.R.; Sasaki, M.; Milman, N.; Keeney, S.; Smith, G.R. Evolutionarily diverse determinants of meiotic DNA break and recombination landscapes across the genome. Genome Res. 2014, 24, 1650–1664. [Google Scholar] [CrossRef] [Green Version]
- Koch, E.N.; Costanzo, M.; Bellay, J.; Deshpande, R.; Chatfield-Reed, K.; Chua, G.; D’Urso, G.; Andrews, B.J.; Boone, C.; Myers, C.L. Conserved rules govern genetic interaction degree across species. Genome Biol. 2012, 13, R57. [Google Scholar] [CrossRef] [Green Version]
- Hurst, L.D.; Pál, C.; Lercher, M.J. The evolutionary dynamics of eukaryotic gene order. Nat. Rev. Genet. 2004, 5, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Tiirikka, T.; Siermala, M.; Vihinen, M. Clustering of gene ontology terms in genomes. Gene 2014, 550, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Acs-Szabo, L.; Papp, L.A.; Miklos, I. Intergenic Sequence Properties of the Schizosaccharomyces (Working Title); Department of Genetics and Applied Microbiology, Faculty of Science and Technology, University of Debrecen: Debrecen, Hungary, 2021; manuscript in preparation. [Google Scholar]
- Dujon, B. Yeast evolutionary genomics. Nat. Rev. Genet. 2010, 11, 512–524. [Google Scholar] [CrossRef]
- Pál, C.; Hurst, L.D. Evidence for co-evolution of gene order and recombination rate. Nat. Genet. 2003, 33, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Shi-Kunne, X.; Faino, L.; van den Berg, G.C.M.; Thomma, B.P.H.J.; Seidl, M.F. Evolution within the fungal genus Verticillium is characterized by chromosomal rearrangement and gene loss. Environ. Microbiol. 2018, 20, 1362–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakirlis, N.; Sarilar, V.; Drillon, G.; Fleiss, A.; Agier, N.; Meyniel, J.P.; Blanpain, L.; Carbone, A.; Devillers, H.; Dubois, K.; et al. Reconstruction of ancestral chromosome architecture and gene repertoire reveals principles of genome evolution in a model yeast genus. Genome Res. 2016, 26, 918–932. [Google Scholar] [CrossRef] [Green Version]
- Burt, D.W.; Bruley, C.; Dunn, I.C.; Jones, C.T.; Ramage, A.; Law, A.S.; Morrice, D.R.; Paton, I.R.; Smith, J.; Windsor, D.; et al. The dynamics of chromosome evolution in birds and mammals. Nature 1999, 402, 411–413. [Google Scholar] [CrossRef]
- Coghlan, A.; Wolfe, K.H. Fourfold faster rate of genome rearrangement in nematodes than in Drosophila. Genome Res. 2002, 12, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Sharakhov, I.V.; Serazin, A.C.; Grushko, O.G.; Dana, A.; Lobo, N.; Hillenmeyer, M.E.; Westerman, R.; Romero-Severson, J.; Costantini, C.; Sagnon, N.; et al. Inversions and gene order shuffling in Anopheles gambiae and A. funestus. Science 2002, 298, 182–185. [Google Scholar] [CrossRef] [Green Version]
- Rajeh, A.; Lv, J.; Lin, Z. Heterogeneous rates of genome rearrangement contributed to the disparity of species richness in Ascomycota. BMC Genom. 2018, 19, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, I.J.; Tanaka, E.; Masuya, H.; Tanaka, R.; Hirooka, Y.; Endoh, R.; Sahashi, N.; Kikuchi, T. Comparative genomics of Taphrina fungi causing varying degrees of tumorous deformity in plants. Genome Biol. Evol. 2014, 6, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.F.; Snell, P.; Gruetzner, F.; Bench, A.J.; Haaf, T.; Metcalfe, J.A.; Green, A.R.; Elgar, G. Analyses of the extent of shared synteny and conserved gene orders between the genome of Fugu rubripes and human 20q. Genome Res. 2002, 12, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Berbee, M.L.; Taylor, J.W. Dating the evolutionary radiations of the true fungi. Can. J. Bot. 1993, 71, 1114–1127. [Google Scholar] [CrossRef]
- Nagy, L.G.; Ohm, R.A.; Kovács, G.M.; Floudas, D.; Riley, R.; Gácser, A.; Sipiczki, M.; Davis, J.M.; Doty, S.L.; de Hoog, G.S.; et al. Latent homology and convergent regulatory evolution underlies the repeated emergence of yeasts. Nat. Commun. 2014, 5, 4471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, S.Y.; Chen, A.X.; Lins, L.S.; Duchêne, D.A.; Lo, N. The Genome as an Evolutionary Timepiece. Genome Biol. Evol. 2016, 8, 3006–3010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, A.; Ho, S.Y.W. The molecular clock and evolutionary timescales. Biochem. Soc. Trans. 2018, 46, 1183–1190. [Google Scholar] [CrossRef]
- Takouridis, S.J.; Tribe, D.E.; Gras, S.L.; Martin, G.J.O. The selective breeding of the freshwater microalga Chlamydomonas reinhardtii for growth in salinity. Bioresour. Technol. 2015, 184, 18–22. [Google Scholar] [CrossRef]
- McDonald, M.J.; Rice, D.P.; Desai, M.M. Sex speeds adaptation by altering the dynamics of molecular evolution. Nature 2016, 531, 233–236. [Google Scholar] [CrossRef] [Green Version]
- Scheuerl, T.; Stelzer, C.P. Sex initiates adaptive evolution by recombination between beneficial loci. PLoS ONE 2017, 12, e0177895. [Google Scholar] [CrossRef]
- Grech, L.; Jeffares, D.C.; Sadée, C.Y.; Rodríguez-López, M.; Bitton, D.A.; Hoti, M.; Biagosch, C.; Aravani, D.; Speekenbrink, M.; Illingworth, C.J.R.; et al. Fitness Landscape of the Fission Yeast Genome. Mol. Biol. Evol. 2019, 36, 1612–1623. [Google Scholar] [CrossRef] [Green Version]
- Naseeb, S.; Carter, Z.; Minnis, D.; Donaldson, I.; Zeef, L.; Delneri, D. Widespread Impact of Chromosomal Inversions on Gene Expression Uncovers Robustness via Phenotypic Buffering. Mol. Biol. Evol. 2016, 33, 1679–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Han, B. Evolutionary conservation of neighbouring gene pairs in plants. Gene 2009, 437, 71–79. [Google Scholar] [CrossRef]
- Dávila López, M.; Martínez Guerra, J.J.; Samuelsson, T. Analysis of gene order conservation in eukaryotes identifies transcriptionally and functionally linked genes. PLoS ONE 2010, 5, e10654. [Google Scholar] [CrossRef]
- Noble, L.M.; Andrianopoulos, A. Fungal genes in context: Genome architecture reflects regulatory complexity and function. Genome Biol. Evol. 2013, 5, 1336–1352. [Google Scholar] [CrossRef] [PubMed]
- Poyatos, J.F.; Hurst, L.D. Is optimal gene order impossible? Trends Genet. 2006, 22, 420–423. [Google Scholar] [CrossRef] [Green Version]
- Al-Shahrour, F.; Minguez, P.; Marqués-Bonet, T.; Gazave, E.; Navarro, A.; Dopazo, J. Selection upon genome architecture: Conservation of functional neighborhoods with changing genes. PLoS Comput. Biol. 2010, 6, e1000953. [Google Scholar] [CrossRef] [Green Version]
- Tuller, T.; Rubinstein, U.; Bar, D.; Gurevitch, M.; Ruppin, E.; Kupiec, M. Higher-order genomic organization of cellular functions in yeast. J. Comput. Biol. 2009, 16, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Tanizawa, H.; Iwasaki, O.; Tanaka, A.; Capizzi, J.R.; Wickramasinghe, P.; Lee, M.; Fu, Z.; Noma, K. Mapping of long-range associations throughout the fission yeast genome reveals global genome organization linked to transcriptional regulation. Nucleic Acids Res. 2010, 38, 8164–8177. [Google Scholar] [CrossRef]
- Gong, K.; Tjong, H.; Zhou, X.J.; Alber, F. Comparative 3D genome structure analysis of the fission and the budding yeast. PLoS ONE 2015, 10, e0119672. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | Number of aLCBs | Number of Genes in aLCBs | Mean Number of Genes in aLCBs | ||||||
| Real Data | 266 | 2055 | 7.73 | ||||||
| Min. | Max. | Mean | Min. | Max. | Mean | Min. | Max. | Mean | |
| Random evolution (n = 100) | 5 | 23 | 13.12 | 33 | 140 | 77.75 | 5.09 | 7.4 | 5.93 |
| ALF evolution (n = 100) | 214 | 289 | 254.54 | 1387 | 1930 | 1656.44 | 6.15 | 6.9 | 6.5 |
| (B) | Number of aLCBs | Number of Genes in aLCBs | Mean Number of Genes in aLCBs | ||||||
| Real vs. Random | Real vs. ALF | Real vs. Random | Real vs. ALF | Real vs. Random | Real vs. ALF | ||||
| Randomisation p | 0.00990099 | 0.188118812 | 0.00990099 | 0.00990099 | 0.00990099 | 0.00990099 | |||
| Single-case t-probe p | 1.53 × 10−83 | 0.44274 | 7.36 × 10−95 | 0.00016125 | 1.24 × 10−5 | 3.33 × 10−14 | |||
| GO Slim Terms | Genes Found Overall | Genes in aLCBs | Genes in NCBs | Randomization p | Single-Case t-Probe p |
|---|---|---|---|---|---|
| Chromatin organization | 275 | 128 | 147 | 0.019608 | 0.043888 |
| Meiotic nuclear division | 142 | 67 | 75 | 0.039216 | 0.072860 |
| Mitochondrial gene expression | 150 | 79 | 71 | 0.019608 | 0.010351 |
| Mitotic sister chromatid segregation | 182 | 91 | 91 | 0.019608 | 0.005432 |
| mRNA metabolic process | 281 | 140 | 141 | 0.019608 | 0.001176 |
| Nucleocytoplasmic transport | 109 | 64 | 45 | 0.019608 | 0.000021 |
| Protein catabolic process | 233 | 114 | 119 | 0.019608 | 0.009971 |
| Protein modification by small protein conjugation or removal | 185 | 89 | 96 | 0.019608 | 0.033979 |
| Protein-containing complex assembly | 262 | 122 | 140 | 0.019608 | 0.030717 |
| Regulation of mitotic cell cycle phase transition | 170 | 80 | 90 | 0.019608 | 0.040627 |
| Ribosome biogenesis | 323 | 156 | 167 | 0.019608 | 0.004532 |
| Transcription, DNA-templated | 459 | 205 | 254 | 0.039216 | 0.044262 |
| tRNA metabolic process | 166 | 93 | 73 | 0.019608 | 0.000877 |
| Vesicle-mediated transport | 319 | 152 | 167 | 0.019608 | 0.000801 |
| Number of Blocks | Number of Genes | Number of Co-Expression Cases | Mean Value of Co-Expression | |
|---|---|---|---|---|
| In aLCBs | 266 | 2055 | 7895 | 0.0408 |
| In NCBs | 232 | 1922 | 8175 | 0.0394 |
| Whole genome | 1 * | 5063 | 12,814,452 | 0.0287 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acs-Szabo, L.; Papp, L.A.; Sipiczki, M.; Miklos, I. Genome Comparisons of the Fission Yeasts Reveal Ancient Collinear Loci Maintained by Natural Selection. J. Fungi 2021, 7, 864. https://doi.org/10.3390/jof7100864
Acs-Szabo L, Papp LA, Sipiczki M, Miklos I. Genome Comparisons of the Fission Yeasts Reveal Ancient Collinear Loci Maintained by Natural Selection. Journal of Fungi. 2021; 7(10):864. https://doi.org/10.3390/jof7100864
Chicago/Turabian StyleAcs-Szabo, Lajos, Laszlo Attila Papp, Matthias Sipiczki, and Ida Miklos. 2021. "Genome Comparisons of the Fission Yeasts Reveal Ancient Collinear Loci Maintained by Natural Selection" Journal of Fungi 7, no. 10: 864. https://doi.org/10.3390/jof7100864