
Single-Cell RNA Sequencing Reveals Distinct Cardiac-Derived Stromal Cell Subpopulations

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials Methods
2.1. CSC Culture and Expansion
2.2. Computational Methods
2.3. Cell Sorting of CSC Subpopulations
3. Results
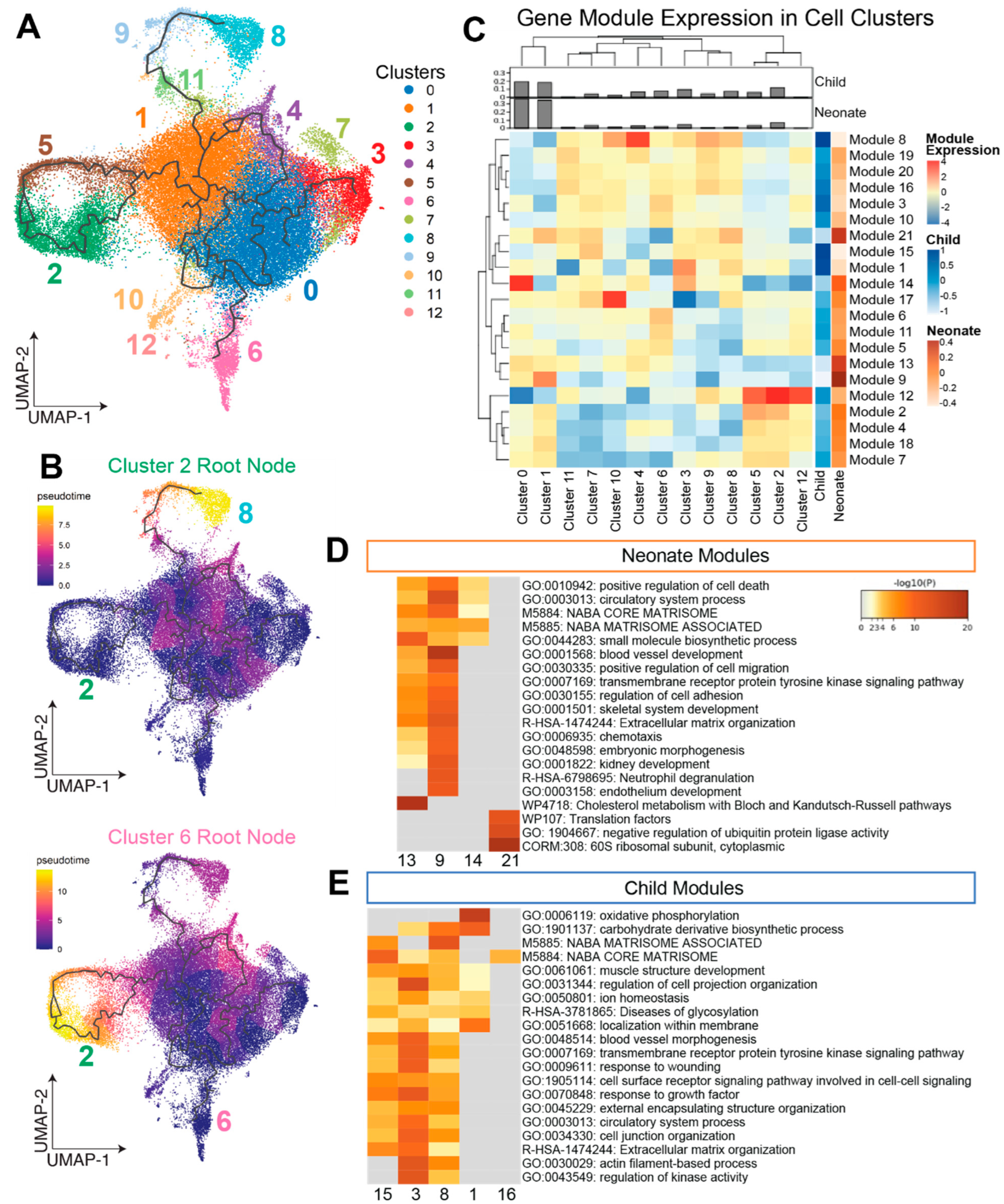
3.1. Clustering and Compositional Analysis Reveal Differences in Neonate and Child CSCs
3.2. Trajectory Analysis Identifies Co-Expressed Genes within CSC Subpopulations
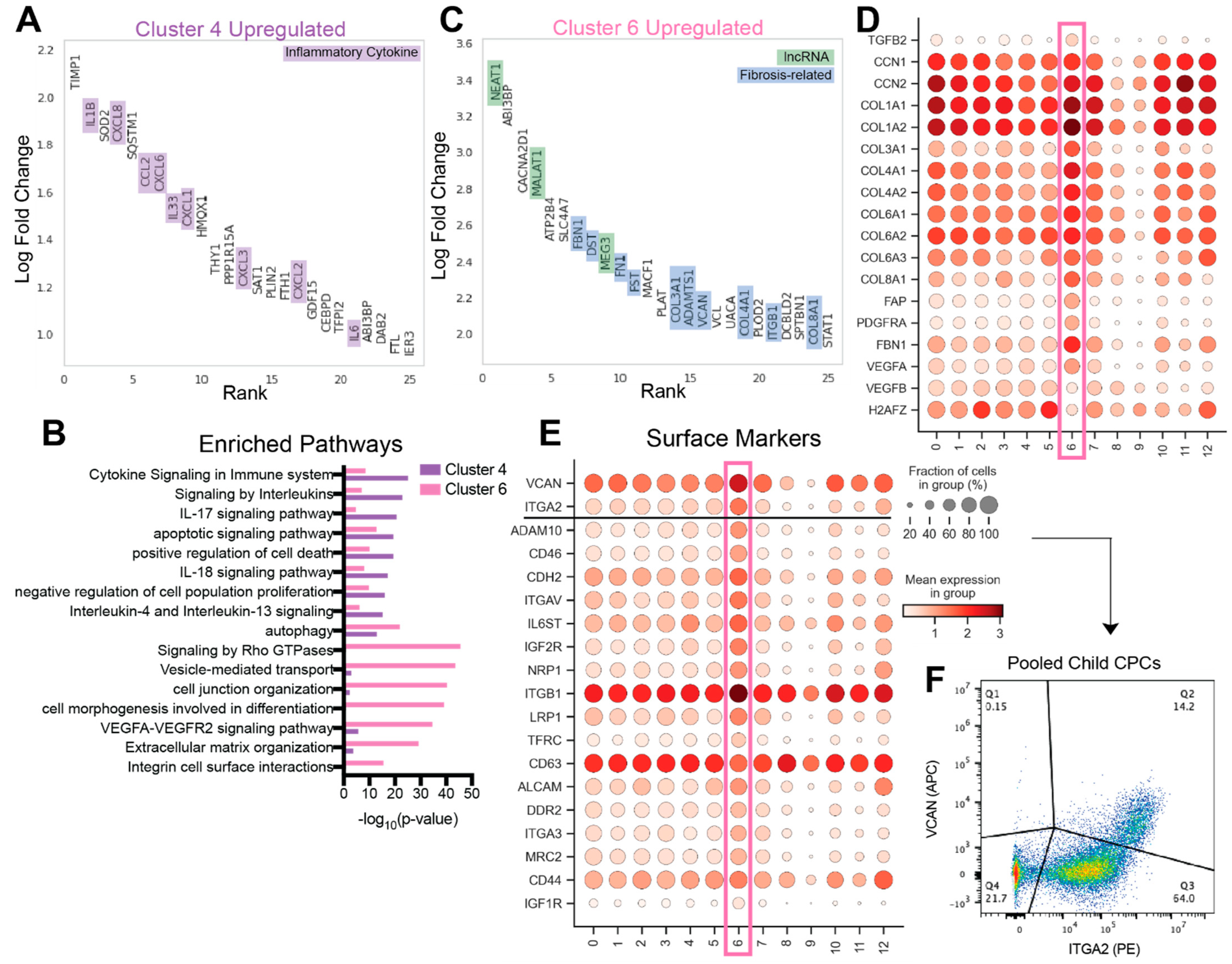
3.3. cCSC-Enriched Cell Clusters 4 and 6 Are Upregulated in Inflammatory Cytokines and Fibrosis-Associated Genes
3.4. Identification of Non-Reparative Surface Markers for Cell Sorting
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Xu, X.-L.; Yi, F.; Pan, H.; Duan, S.; Ding, Z.-C.; Yuan, G.-H.; Qu, J.; Zhang, H.; Liu, G.-H. Progress and prospects in stem cell therapy. Acta Pharmacol. Sin. 2013, 34, 741–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, C.; Brindley, D.A.; Culme-Seymour, E.J.; Davie, N.L. Cell therapy industry: Billion dollar global business with unlimited potential. Regen. Med. 2011, 6, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Shoja-Taheri, F.; George, A.; Agarwal, U.; Platt, M.O.; Gibson, G.; Davis, M.E. Using Statistical Modeling to Understand and Predict Pediatric Stem Cell Function. Circ. Genom. Precis. Med. 2019, 12, e002403. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, U.; Smith, A.W.; French, K.M.; Boopathy, A.V.; George, A.; Trac, D.; Brown, M.E.; Shen, M.; Jiang, R.; Fernandez, J.D.; et al. Age-Dependent Effect of Pediatric Cardiac Progenitor Cells after Juvenile Heart Failure. Stem Cells Transl. Med. 2016, 5, 883–892. [Google Scholar] [CrossRef]
- Sharma, S.; Mishra, R.; Bigham, G.E.; Wehman, B.; Khan, M.M.; Xu, H.; Saha, P.; Goo, Y.A.; Datla, S.R.; Chen, L.; et al. A Deep Proteome Analysis Identifies the Complete Secretome as the Functional Unit of Human Cardiac Progenitor Cells. Circ. Res. 2017, 120, 816–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efimenko, A.; Dzhoyashvili, N.; Kalinina, N.; Kochegura, T.; Akchurin, R.; Tkachuk, V.; Parfyonova, Y. Adipose-Derived Mesenchymal Stromal Cells from Aged Patients with Coronary Artery Disease Keep Mesenchymal Stromal Cell Properties but Exhibit Characteristics of Aging and Have Impaired Angiogenic Potential. Stem Cells Transl. Med. 2014, 3, 32–41. [Google Scholar] [CrossRef]
- Jumabay, M.; Moon, J.H.; Yeerna, H.; Boström, K.I. Effect of Diabetes Mellitus on Adipocyte-Derived Stem Cells in Rat. J. Cell. Physiol. 2015, 230, 2821–2828. [Google Scholar] [CrossRef] [Green Version]
- Stolzing, A.; Jones, E.; McGonagle, D.; Scutt, A. Age-related changes in human bone marrow-derived mesenchymal stem cells: Consequences for cell therapies. Mech. Ageing Dev. 2008, 129, 163–173. [Google Scholar] [CrossRef]
- Khong, S.M.L.; Lee, M.; Kosaric, N.; Khong, D.M.; Dong, Y.; Hopfner, U.; Aitzetmüller, M.M.; Duscher, D.; Schäfer, R.; Gurtner, G.C. Single-Cell Transcriptomics of Human Mesenchymal Stem Cells Reveal Age-Related Cellular Subpopulation Depletion and Impaired Regenerative Function. Stem Cells 2019, 37, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.H.; Han, J.W.; Lee, J.Y.; Choi, Y.J.; Sohn, Y.-D.; Song, M.; Yoon, Y.-S. Diabetic Mesenchymal Stem Cells Are Ineffective for Improving Limb Ischemia Due to Their Impaired Angiogenic Capability. Cell Transplant. 2015, 24, 1571–1584. [Google Scholar] [CrossRef]
- Fan, M.; Chen, W.; Liu, W.; Du, G.-Q.; Jiang, S.-L.; Tian, W.-C.; Sun, L.; Li, R.-K.; Tian, H. The Effect of Age on the Efficacy of Human Mesenchymal Stem Cell Transplantation after a Myocardial Infarction. Rejuvenation Res. 2010, 13, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Vasa, M.; Fichtlscherer, S.; Aicher, A.; Adler, K.; Urbich, C.; Martin, H.; Zeiher, A.M.; Dimmeler, S. Number and Migratory Activity of Circulating Endothelial Progenitor Cells Inversely Correlate with Risk Factors for Coronary Artery Disease. Circ. Res. 2001, 89, E1–E7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Yu, W.; Ye, G.; Li, J.; Zheng, G.; Liu, W.; Lin, J.; Su, Z.; Che, Y.; Ye, F.; et al. Single-cell RNA sequencing analysis of human bone-marrow-derived mesenchymal stem cells and functional subpopulation identification. Exp. Mol. Med. 2022, 54, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Han, G.; Puebla-Osorio, N.; Ma, M.C.J.; Strati, P.; Chasen, B.; Dai, E.; Dang, M.; Jain, N.; Yang, H.; et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med. 2020, 26, 1878–1887. [Google Scholar] [CrossRef]
- Sheih, A.; Voillet, V.; Hanafi, L.-A.; DeBerg, H.A.; Yajima, M.; Hawkins, R.; Gersuk, V.; Riddell, S.R.; Maloney, D.G.; Wohlfahrt, M.E.; et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun. 2020, 11, 219. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, S.; Hare, J.M.; Shah, A.M.; Pietris, N.P.; Bettencourt, J.L.; Piller, L.B.; Khan, A.; Snyder, A.; Boyd, R.M.; Abdullah, M.; et al. Autologous Cardiac Stem Cell Injection in Patients with Hypoplastic Left Heart Syndrome (CHILD Study). Pediatr. Cardiol. 2022, 43, 1481–1493. [Google Scholar] [CrossRef]
- Saha, P.; Sharma, S.; Korutla, L.; Datla, S.R.; Shoja-Taheri, F.; Mishra, R.; Bigham, G.E.; Sarkar, M.; Morales, D.; Bittle, G.; et al. Circulating exosomes derived from transplanted progenitor cells aid the functional recovery of ischemic myocardium. Sci. Transl. Med. 2019, 11, eaau1168. [Google Scholar] [CrossRef]
- Tang, X.-L.; Rokosh, D.G.; Guo, Y.; Bolli, R. Cardiac Progenitor Cells and Bone Marrow-Derived Very Small Embryonic-Like Stem Cells for Cardiac Repair after Myocardial Infarction. Circ. J. 2010, 74, 390–404. [Google Scholar] [CrossRef] [Green Version]
- Bolli, R.; Mitrani, R.D.; Hare, J.M.; Pepine, C.J.; Perin, E.C.; Willerson, J.T.; Traverse, J.H.; Henry, T.D.; Yang, P.C.; Murphy, M.P.; et al. A Phase II study of autologous mesenchymal stromal cells and c-kit positive cardiac cells, alone or in combination, in patients with ischaemic heart failure: The CCTRN CONCERT-HF trial. Eur. J. Heart Fail. 2021, 23, 661–674. [Google Scholar] [CrossRef]
- Tang, Y.; Zhu, W.; Cheng, M.; Chen, L.; Zhang, J.; Sun, T.; Kishore, R.; Phillips, M.I.; Losordo, D.; Qin, G. Hypoxic Preconditioning Enhances the Benefit of Cardiac Progenitor Cell Therapy for Treatment of Myocardial Infarction by Inducing CXCR4 Expression. Circ. Res. 2009, 104, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Yao, Y.; Chen, L.; Li, Y.; Sheng, Z.; Ma, G. Hypoxic Preconditioning Improves Survival of Cardiac Progenitor Cells: Role of Stromal Cell Derived Factor-1α–CXCR4 Axis. PLoS ONE 2012, 7, e37948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, I.; Baio, J.M.; Tsay, E.; Martinez, A.F.; Fuentes, T.I.; Bailey, L.L.; Hasaniya, N.W.; Kearns-Jonker, M. Short-term hypoxia improves early cardiac progenitor cell function in vitro. Am. J. Stem Cells 2018, 7, 1–17. [Google Scholar]
- Trac, D.; Maxwell, J.T.; Brown, M.E.; Xu, C.; Davis, M.E. Aggregation of Child Cardiac Progenitor Cells Into Spheres Activates Notch Signaling and Improves Treatment of Right Ventricular Heart Failure. Circ. Res. 2019, 124, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, M.; Mishra, R.; Saha, P.; Morales, D.; Cheng, W.-C.; Jayaraman, A.R.; Hoffman, J.R.; Davidson, L.; Chen, L.; Shah, A.M.; et al. Comparative efficacy and mechanism of action of cardiac progenitor cells after cardiac injury. iScience 2022, 25, 104656. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, T.I.; Appleby, N.; Tsay, E.; Martinez, J.J.; Bailey, L.; Hasaniya, N.; Kearns-Jonker, M. Human Neonatal Cardiovascular Progenitors: Unlocking the Secret to Regenerative Ability. PLoS ONE 2013, 8, e77464. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Vijayan, K.; Colletti, E.J.; Harrington, D.A.; Matthiesen, T.S.; Simpson, D.; Goh, S.K.; Walker, B.L.; Almeida-Porada, G.; Wang, D.; et al. Characterization and Functionality of Cardiac Progenitor Cells in Congenital Heart Patients. Circulation 2011, 123, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [Green Version]
- Wolock, S.L.; Lopez, R.; Klein, A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019, 8, 281–291.e9. [Google Scholar] [CrossRef] [Green Version]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat. Commun. 2022, 13, 1246. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.H.; Simonds, E.F.; Bendall, S.C.; Davis, K.L.; Amir, E.-A.D.; Tadmor, M.D.; Litvin, O.; Fienberg, H.G.; Jager, A.; Zunder, E.R.; et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haghverdi, L.; Lun, A.T.L.; Morgan, M.D.; Marioni, J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018, 36, 421–427. [Google Scholar] [CrossRef]
- Traag, V.A.; Waltman, L.; Van Eck, N.J. From Louvain to Leiden: Guaranteeing well-connected communities. Sci. Rep. 2019, 9, 5233. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.A.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Bausch-Fluck, D.; Hofmann, A.; Bock, T.K.C.; Frei, A.P.; Cerciello, F.; Jacobs, A.; Moest, H.; Omasits, U.; Gundry, R.L.; Yoon, C.; et al. A Mass Spectrometric-Derived Cell Surface Protein Atlas. PLoS ONE 2015, 10, e0121314. [Google Scholar] [CrossRef] [Green Version]
- Bouzeghrane, F.; Reinhardt, D.; Reudelhuber, T.L.; Thibault, G. Enhanced expression of fibrillin-1, a constituent of the myocardial extracellular matrix in fibrosis. Am. J. Physiol. Circ. Physiol. 2005, 289, H982–H991. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Wang, Y.; Zhang, W.; Jia, Q.; Wang, X.; Li, Y.; Lv, S.; Zhang, J. Roles of Biomarkers in Myocardial Fibrosis. Aging Dis. 2020, 11, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Kahounová, Z.; Kurfurstova, D.; Bouchal, J.; Kharaishvili, G.; Navrátil, J.; Remšík, J.; Šimečková, S.; Študent, V.; Kozubík, A.; Souček, K. The fibroblast surface markers FAP, anti-fibroblast, and FSP are expressed by cells of epithelial origin and may be altered during epithelial-to-mesenchymal transition. Cytom. Part A 2018, 93, 941–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jechlinger, M.; Sommer, A.; Moriggl, R.; Seither, P.; Kraut, N.; Capodiecci, P.; Donovan, M.; Cordon-Cardo, C.; Beug, H.; Grünert, S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Investig. 2006, 116, 1561–1570. [Google Scholar] [CrossRef]
- Ge, Z.; Yin, C.; Li, Y.; Tian, D.; Xiang, Y.; Li, Q.; Tang, Y.; Zhang, Y. Long noncoding RNA NEAT1 promotes cardiac fibrosis in heart failure through increased recruitment of EZH2 to the Smad7 promoter region. J. Transl. Med. 2022, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhang, L.; Song, J.; Wang, Z.; Huang, X.; Guo, Z.; Chen, F.; Zhao, X. Long noncoding RNA MALAT1 mediates cardiac fibrosis in experimental postinfarct myocardium mice model. J. Cell. Physiol. 2019, 234, 2997–3006. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Ma, X.; Lin, S.; Wu, B.; Chen, Y.; Peng, C. Long noncoding RNA-MEG3 contributes to myocardial ischemia–reperfusion injury through suppression of miR-7-5p expression. Biosci. Rep. 2019, 39, BSR20190210. [Google Scholar] [CrossRef] [Green Version]
- Konstandin, M.H.; Toko, H.; Gastelum, G.M.; Quijada, P.; De La Torre, A.; Quintana, M.; Collins, B.; Din, S.; Avitabile, D.; Völkers, M.; et al. Fibronectin Is Essential for Reparative Cardiac Progenitor Cell Response After Myocardial Infarction. Circ. Res. 2013, 113, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, J.R.; Park, H.-J.; Bheri, S.; Jayaraman, A.R.; Davis, M.E. Comparative computational RNA analysis of cardiac-derived progenitor cells and their extracellular vesicles. Genomics 2022, 114, 110349. [Google Scholar] [CrossRef]
- Vagnozzi, R.J.; Maillet, M.; Sargent, M.A.; Khalil, H.; Johansen, A.K.Z.; Schwanekamp, J.A.; York, A.J.; Huang, V.; Nahrendorf, M.; Sadayappan, S.; et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 2020, 577, 405–409. [Google Scholar] [CrossRef]
- Van Berlo, J.H.; Kanisicak, O.; Maillet, M.; Vagnozzi, R.J.; Karch, J.; Lin, S.-C.J.; Middleton, R.C.; Marbán, E.; Molkentin, J.D. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 2014, 509, 337–341. [Google Scholar] [CrossRef] [Green Version]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Lelek, S.; Spanjaard, B.; El-Sammak, H.; Simões, M.G.; Mintcheva, J.; Aliee, H.; Schäfer, R.; Meyer, A.M.; Theis, F.; et al. Origin and function of activated fibroblast states during zebrafish heart regeneration. Nat. Genet. 2022, 54, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.R.; Ranjbarvaziri, S.; Talkhabi, M.; Zhao, P.; Subat, A.; Hojjat, A.; Kamran, P.; Müller, A.M.; Volz, K.S.; Tang, Z.; et al. Developmental Heterogeneity of Cardiac Fibroblasts Does Not Predict Pathological Proliferation and Activation. Circ. Res. 2014, 115, 625–635. [Google Scholar] [CrossRef] [Green Version]
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.; E Nordon, R.; Harvey, R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. eLife 2019, 8, e43882. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffman, J.R.; Jayaraman, A.R.; Bheri, S.; Davis, M.E. Single-Cell RNA Sequencing Reveals Distinct Cardiac-Derived Stromal Cell Subpopulations. J. Cardiovasc. Dev. Dis. 2022, 9, 374. https://doi.org/10.3390/jcdd9110374
Hoffman JR, Jayaraman AR, Bheri S, Davis ME. Single-Cell RNA Sequencing Reveals Distinct Cardiac-Derived Stromal Cell Subpopulations. Journal of Cardiovascular Development and Disease. 2022; 9(11):374. https://doi.org/10.3390/jcdd9110374
Chicago/Turabian StyleHoffman, Jessica R., Arun R. Jayaraman, Sruti Bheri, and Michael E. Davis. 2022. "Single-Cell RNA Sequencing Reveals Distinct Cardiac-Derived Stromal Cell Subpopulations" Journal of Cardiovascular Development and Disease 9, no. 11: 374. https://doi.org/10.3390/jcdd9110374