
The Pharmacokinetic and Absolute Bioavailability of Cyclosporine (Atopica for Cats®) in Cats
 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Background
2. Materials and Methods
2.1. Chemical and Reagents
2.2. Standards Solutions
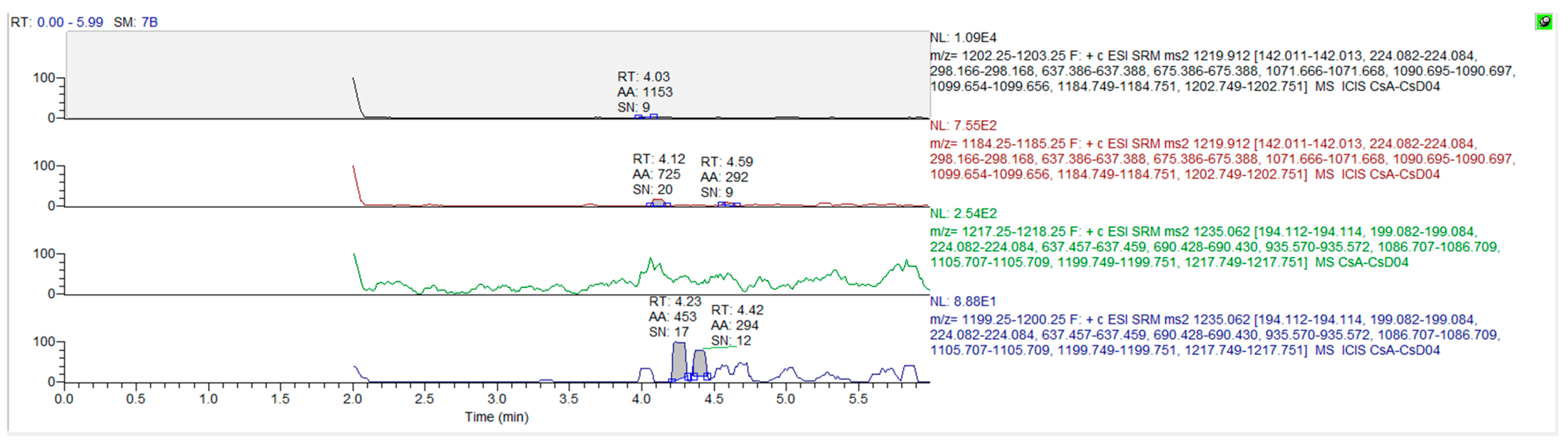
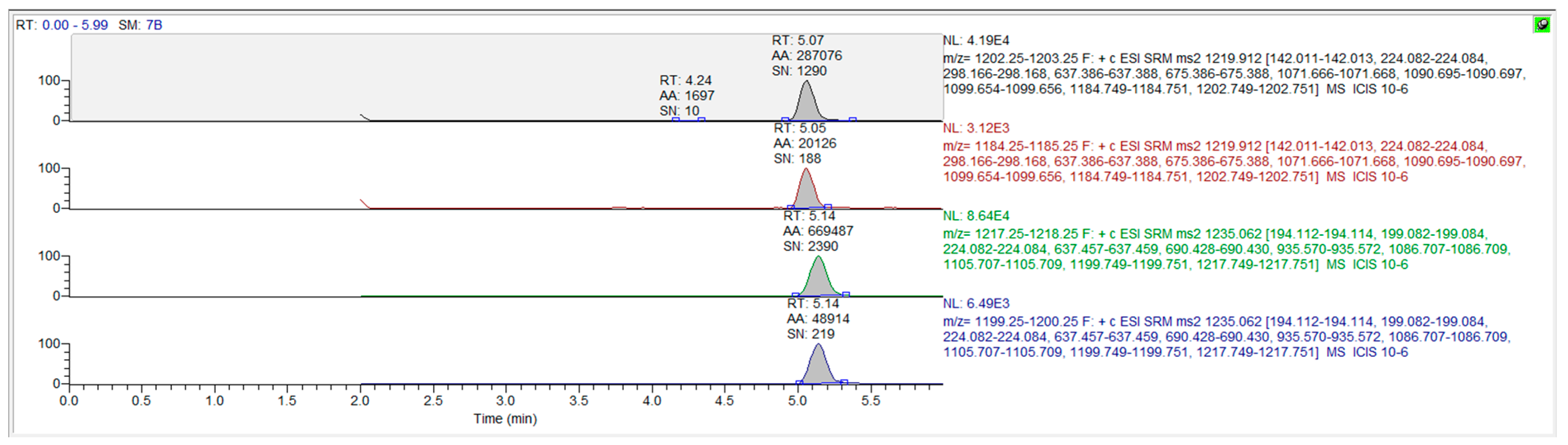
2.3. Method Establishment and Validation
2.4. Animals
2.5. Study Design
2.6. Sample Preparation
2.7. Data Analysis
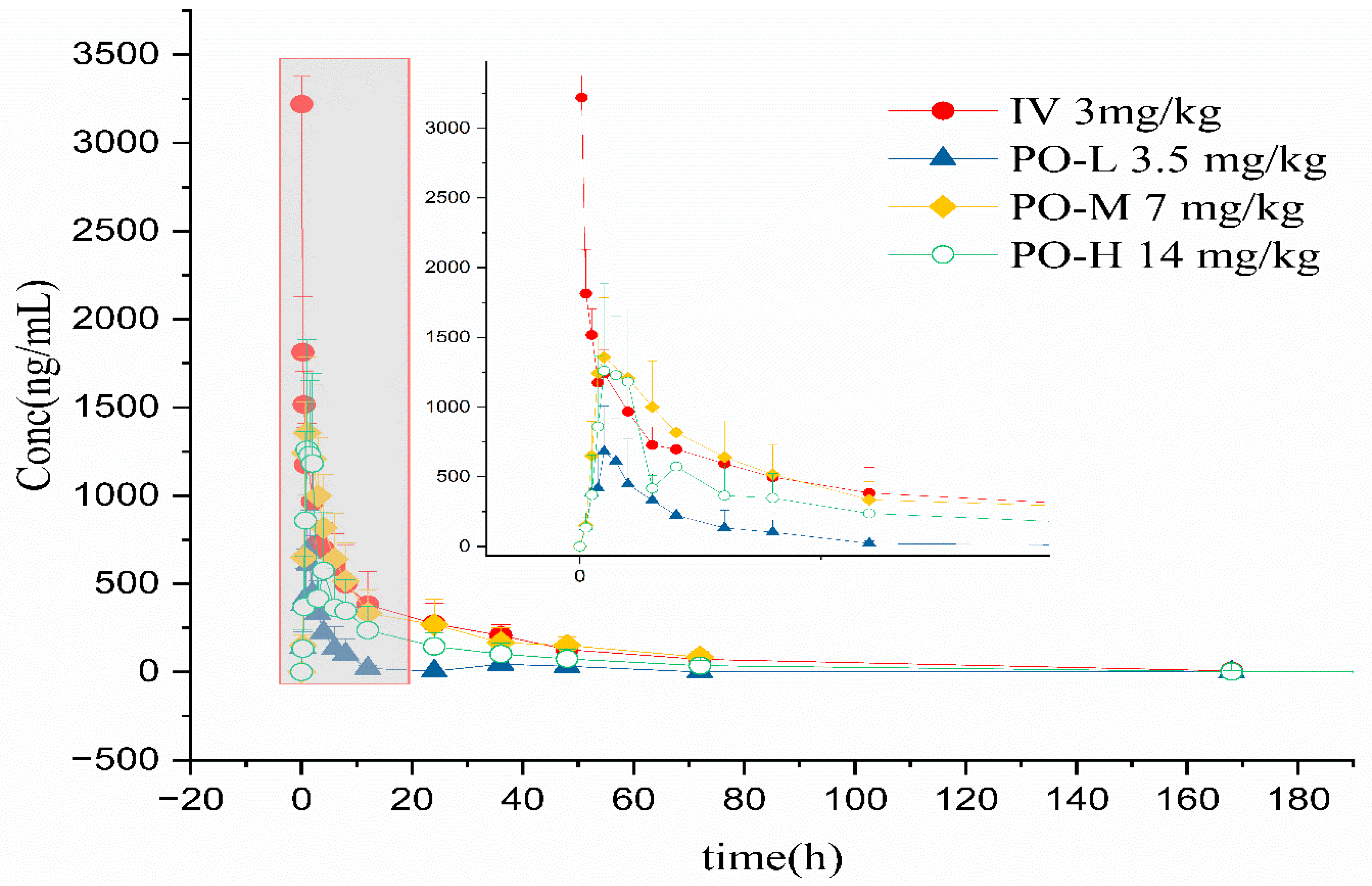
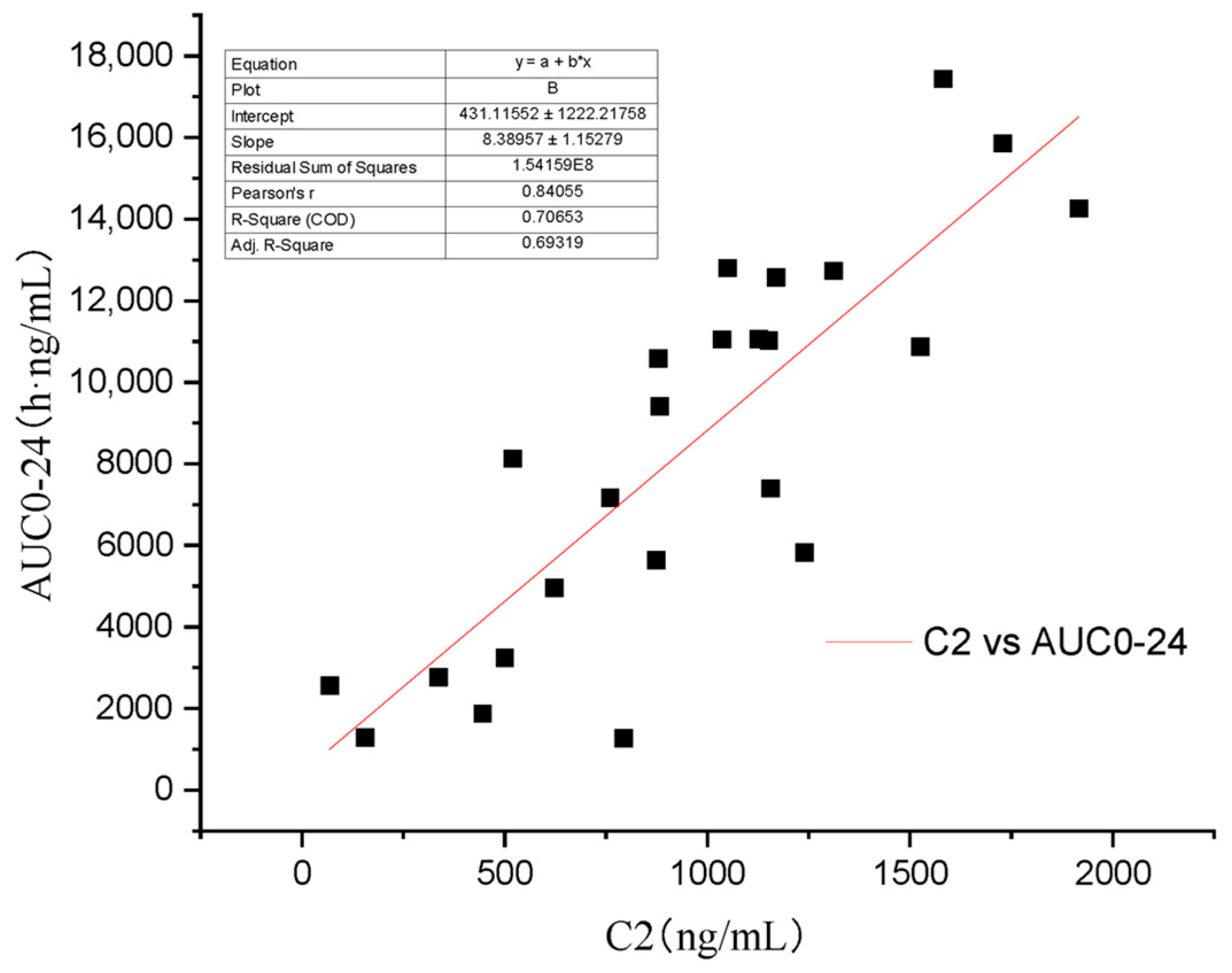
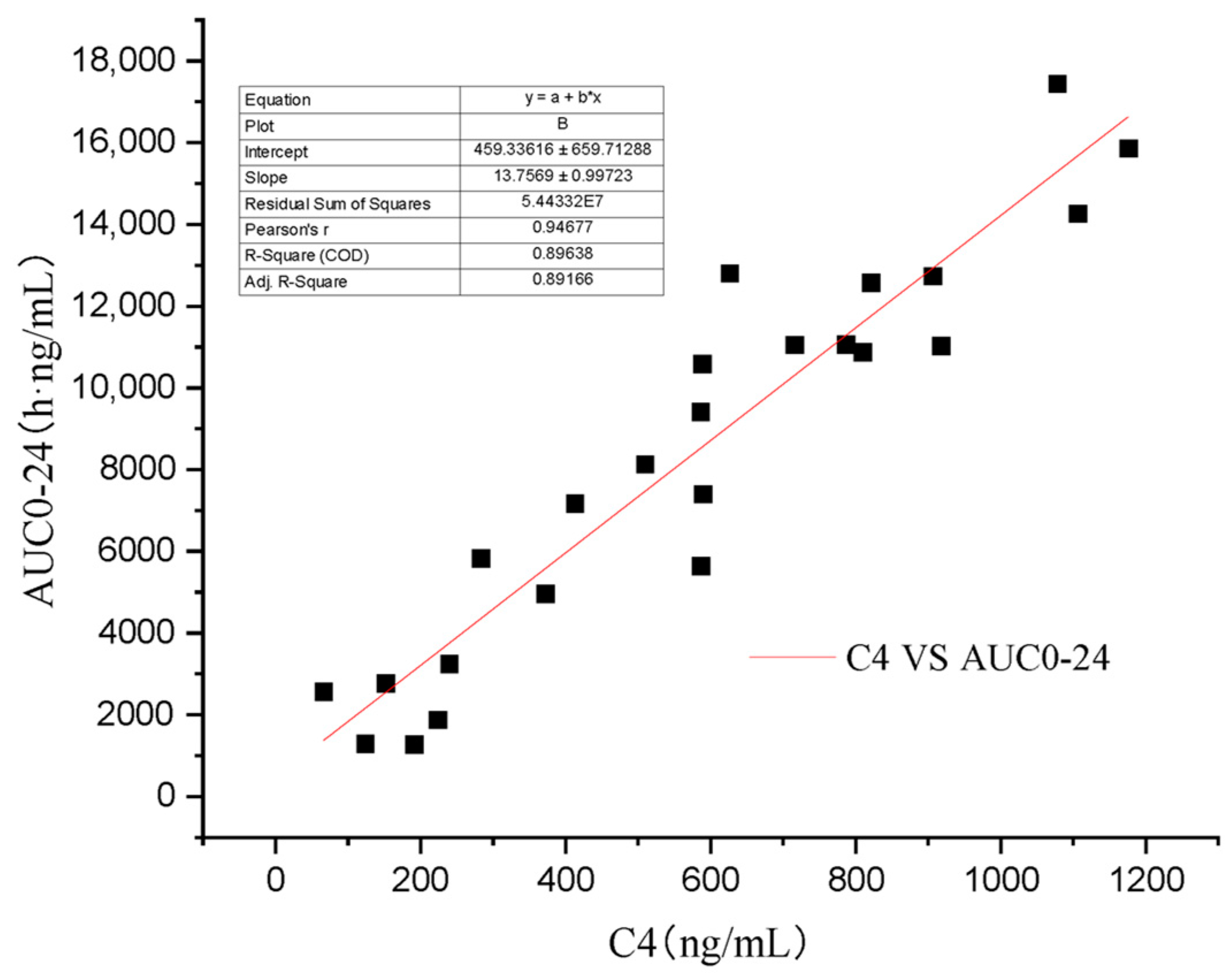
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kovalik, M.; Thoday, K.L.; van den Broek, A.H. The use of ciclosporin A in veterinary dermatology. Vet. J. 2012, 193, 317–325. [Google Scholar] [CrossRef]
- Robson, D. Review of the pharmacokinetics, interactions and adverse reactions of cyclosporine in people, dogs and cats. Vet. Rec. 2003, 152, 739–748. [Google Scholar] [CrossRef]
- Mohamed, R.; Mercolini, L.; Cuennet-Cosandey, S.; Chavent, J.; Raggi, M.A.; Peyrou, M. Validation of a dried blood spot LC-MS/MS approach for cyclosporin A in cat blood: Comparison with a classical sample preparation. J. Pharm. Biomed. Anal. 2012, 66, 298–305. [Google Scholar] [CrossRef]
- Colombo, S.; Sartori, R. Ciclosporin and the cat: Current understanding and review of clinical use. J. Feline Med. Surg. 2018, 20, 244–255. [Google Scholar] [CrossRef]
- Cridge, H.; Kordon, A.; Pinchuk, L.M.; Wills, R.W.; Thomason, J.M.; Mackin, A.J.; Archer, T.M. Effects of cyclosporine on feline lymphocytes activated in vitro. Vet. Immunol. Immunopathol. 2020, 219, 109962. [Google Scholar] [CrossRef]
- Masri, M.A.; Barbari, A.; Stephan, A.; Rizk, S.; Kilany, H.; Kamel, G. Measurement of lymphocyte cyclosporine levels in transplant patients. Transplant. Proc. 1998, 30, 3561–3562. [Google Scholar] [CrossRef] [PubMed]
- Wisselink, M.A.; Willemse, T. The efficacy of cyclosporine A in cats with presumed atopic dermatitis: A double blind, randomised prednisolone-controlled study. Vet. J. 2009, 180, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Russell, G.; Graveley, R.; Seid, J.; al-Humidan, A.K.; Skjodt, H. Mechanisms of action of cyclosporine and effects on connective tissues. Semin. Arthritis Rheum. 1992, 21, 16–22. [Google Scholar] [CrossRef]
- Marsella, R. Atopic Dermatitis in Domestic Animals: What Our Current Understanding Is and How This Applies to Clinical Practice. Vet. Sci. 2021, 8, 124. [Google Scholar] [CrossRef]
- Palmeiro, B.S. Cyclosporine in veterinary dermatology. Vet. Clin. N. Am. Small Anim. Pract. 2013, 43, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Fradette, C.; Lavigne, J.; Waters, D.; Ducharme, M.P. The utility of the population approach applied to bioequivalence in patients: Comparison of 2 formulations of cyclosporine. Ther. Drug Monit. 2005, 27, 592–600. [Google Scholar] [CrossRef]
- Archer, T.M.; Boothe, D.M.; Langston, V.C.; Fellman, C.L.; Lunsford, K.V.; Mackin, A.J. Oral cyclosporine treatment in dogs: A review of the literature. J. Vet. Intern. Med. 2014, 28, 1–20. [Google Scholar] [CrossRef]
- FDA. ATOPICA for Cats (Cyclosporine Oral Solution) USP MODIFIED. Available online: https://animaldrugsatfda.fda.gov/adafda/app/search/public/document/downloadLabeling/881 (accessed on 25 June 2020).
- FDA. Freedom of Information Summary. Original Abbreviated New Animal Drug Application. Available online: https://animaldrugsatfda.fda.gov/adafda/app/search/public/document/downloadFoi/13676 (accessed on 29 March 2023).
- Lappin, M.R.; VanLare, K.A.; Seewald, W.; Roycroft, L.M.; Scorza, A.V.; King, S.; Roberts, E.S. Effect of oral administration of cyclosporine on Toxoplasma gondii infection status of cats. Am. J. Vet. Res. 2015, 76, 351–357. [Google Scholar] [CrossRef]
- Salant, H.; Klainbart, S.; Kelmer, E.; Mazuz, M.L.; Baneth, G.; Aroch, I. Systemic toxoplasmosis in a cat under cyclosporine therapy. Vet. Parasitol. Reg. Stud. Rep. 2021, 23, 100542. [Google Scholar] [CrossRef]
- Last, R.D.; Suzuki, Y.; Manning, T.; Lindsay, D.; Galipeau, L.; Whitbread, T.J. A case of fatal systemic toxoplasmosis in a cat being treated with cyclosporin A for feline atopy. Vet. Dermatol. 2004, 15, 194–198. [Google Scholar] [CrossRef]
- Favrot, C. Feline non-flea induced hypersensitivity dermatitis: Clinical features, diagnosis and treatment. J. Feline Med. Surg. 2013, 15, 778–784. [Google Scholar] [CrossRef]
- FDA. Freedom of Information Summary-Original New Animal Drug Application-Atopica for Cats (Cyclosporine Oral Solution, USP) Modified Cats. Available online: https://animaldrugsatfda.fda.gov/adafda/app/search/public/document/downloadFoi/888 (accessed on 8 August 2011).
- Cheng, H.Y.; Jusko, W.J. Application of mean residence-time concepts to pharmacokinetic systems with noninstantaneous input and nonlinear elimination. Pharm. Res. 1989, 6, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Mehl, M.L.; Kyles, A.E.; Craigmill, A.L.; Epstein, S.; Gregory, C.R. Disposition of cyclosporine after intravenous and multi-dose oral administration in cats. J. Vet. Pharmacol. Ther. 2003, 26, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kong, J.; Liu, Y.; Wu, Q.; Cao, Y.; Qiu, J.; Zhang, L.; Gong, X.; Zhao, F.; Cao, X.; et al. Pharmacokinetics and bioequivalence of two cyclosporine oral solution formulations in cats. Front. Vet. Sci 2022, 9, 940472. [Google Scholar] [CrossRef]
- Lai, J.; Lu, Y.; Yin, Z.; Hu, F.; Wu, W. Pharmacokinetics and enhanced oral bioavailability in beagle dogs of cyclosporine A encapsulated in glyceryl monooleate/poloxamer 407 cubic nanoparticles. Int. J. Nanomed. 2010, 5, 13–23. [Google Scholar]
- Mehvar, R. Principles of nonlinear pharmacokinetics. Am. J. Pharm. Educ. 2001, 65, 178–184. [Google Scholar]
- Barr, W.H. Cyclosporine: The case for expanding bioequivalence criteria to include measures of individual bioequivalence in relevant population subsets. Transplant. Proc. 1999, 31, 25s–30s. [Google Scholar] [CrossRef] [PubMed]
- McAnulty, J.F.; Lensmeyer, G.L. The effects of ketoconazole on the pharmacokinetics of cyclosporine A in cats. Vet. Surg. 1999, 28, 448–455. [Google Scholar] [CrossRef]
- Holm, N.B.; Deryabina, M.; Knudsen, C.B.; Janfelt, C. Tissue distribution and metabolic profiling of cyclosporine (CsA) in mouse and rat investigated by DESI and MALDI mass spectrometry imaging (MSI) of whole-body and single organ cryo-sections. Anal. Bioanal. Chem. 2022, 414, 7167–7177. [Google Scholar] [CrossRef] [PubMed]
- Albitar, O.; Ballouze, R.; Harun, S.N.; Mohamed Noor, D.A.; Sheikh Ghadzi, S.M. Population Pharmacokinetic Modeling of Cyclosporine Among Malaysian Renal Transplant Patients: An Evaluation of Methods to Handle Missing Doses in Conventional Drug-Monitoring Data. J. Clin. Pharmacol. 2020, 60, 1474–1482. [Google Scholar] [CrossRef]
- Umpierrez, M.; Guevara, N.; Ibarra, M.; Fagiolino, P.; Vazquez, M.; Maldonado, C. Development of a Population Pharmacokinetic Model for Cyclosporine from Therapeutic Drug Monitoring Data. Biomed. Res. Int. 2021, 2021, 3108749. [Google Scholar] [CrossRef]
- Katayama, M.; Katayama, R.; Kamishina, H. Effects of multiple oral dosing of itraconazole on the pharmacokinetics of cyclosporine in cats. J. Feline Med. Surg. 2010, 12, 512–514. [Google Scholar] [CrossRef]
- Marsella, R.; De Benedetto, A. Atopic Dermatitis in Animals and People: An Update and Comparative Review. Vet. Sci. 2017, 4, 37. [Google Scholar] [CrossRef]
- Kuilenburg, A.B.V.; Maring, J.G. Evaluation of 5-fluorouracil pharmacokinetic models and therapeutic drug monitoring in cancer patients. Pharmacogenomics 2013, 14, 799–811. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Intravenous Group |
|---|---|
| tmax (h) | 0.17 ± 0.19 |
| Cmax (μg/mL) | 2.91 ± 0.62 |
| t1/2 (h) | 30.3 ± 7.25 |
| AUC0–t (h’μg/mL) | 21.6 ± 3.53 |
| AUC0–∞ (h´μg/mL) | 22.0 ± 3.46 |
| Vd (L/kg) | 6.21 ± 2.22 |
| CL (L/h/kg) | 0.140 ± 0.0252 |
| MRT0–t (h) | 31.9 ± 2.04 |
| AUC0–4 (h´μg/mL) | 4.12 ± 0.309 |
| AUC0–24 (h´μg/mL) | 11.0 ± 1.88 |
| AUC/Dose | 7.33 ± 1.15 |
| Vss_obs (L) | 4930 ± 1230 |
| Parameters | Low Oral Group | Medium Oral Group | High Oral Group |
|---|---|---|---|
| tmax (h) | 1.08 ± 0.38 | 0.92 ± 0.13 | 1.42 ± 0.38 |
| Cmax (μg/mL) | 0.778 ± 0.269 | 1.39 ± 0.381 | 1.38 ± 0.571 |
| t1/2 (h) | 8.06 ± 4.45 | 25.8 ± 10.2 | 20.1 ± 7.67 |
| AUC0–t (h´μg/mL) | 3.69 ± 2.58 | 18.7 ± 8.24 | 13.6 ± 7.87 |
| AUC0–∞ (h´μg/mL) | 3.74 ± 2.58 | 21.9 ± 10.6 | 13.9 ± 7.69 |
| Vd/F (L/kg) | 38.4 ± 42.3 | 25.0 ± 3.12 | 35.3 ± 17.0 |
| CL/F (L/h/kg) | 2.76 ± 1.82 | 0.921 ± 0.803 | 1.47 ± 1.13 |
| MRT0–t (h) | 13.2 ± 6.98 | 18.9 ± 5.47 | 22.6 ± 8.69 |
| AUC0–4 (h´μg/mL) | 1.57 ± 0.816 | 3.97 ± 1.29 | 3.02 ± 1.14 |
| AUC0–24 (h´μg/mL) | 2.57 ± 1.63 | 11.9 ± 4.52 | 8.13 ± 3.89 |
| AUC0-∞/Dose | 1.07 ± 0.737 | 3.13 ± 1.51 | 0.993 ± 0.549 |
| Parameters | Intravenous Group | Low Oral Group | Medium Oral Group | High Oral Group |
|---|---|---|---|---|
| A | 1170 ± 127 | −312,000 ± 542,000 | 79,200,000 ± 144,000,000 | −0.38 ± 0.36 |
| B | 479 ± 292 | 216 ± 451 | 557 ± 201 | 2220 ± 919 |
| α | 0.45 ± 0.27 | 1.17 ± 1.26 | 0.75 ± 0.08 | 2.49 ± 0.02 |
| β | 0.02 ± 0.01 | 0.42 ± 0.14 | 0.04 ± 0.02 | 0.24 ± 0.07 |
| t1/2 α (h) | 2.12 ± 1.39 | 1.07 ± 0.69 | 0.93 ± 0.08 | 0.28 ± 0.00 |
| t1/2 β (h) | 36.1 ± 22.5 | 1.81 ± 0.61 | 21.3 ± 5.68 | 3.06 ± 0.80 |
| t1/2 e (h) | 10.2 ± 3.19 | 1.16 ± 1.27 | 4.52 ± 1.46 | 3.06 ± 0.80 |
| K12 | 0.23 ± 0.11 | 0.00 ± 0.00 | 0.45 ± 0.02 | 0.00 ± 0.00 |
| K21 | 0.16 ± 0.16 | 0.43 ± 0.14 | 0.16 ± 0.02 | 2.49 ± 0.02 |
| Ke | 0.07 ± 0.02 | 1.16 ± 1.27 | 0.18 ± 0.09 | 0.24 ± 0.07 |
| t1/2 Ka (h) | 0.93 ± 0.28 | 0.93 ± 0.08 | 0.51 ± 0.10 |
| Models | Estimate | F-Stat | p-Value | Lower CI (95%) | Upper CI (95%) |
|---|---|---|---|---|---|
| Power model | 0.96 | 0.24 | 1.69 | ||
| One Way ANOVA | 10.98 | 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, J.; Yang, Y.; Liu, Y.; Cao, Y.; Qiu, J.; Sun, P.; Cao, X. The Pharmacokinetic and Absolute Bioavailability of Cyclosporine (Atopica for Cats®) in Cats. Vet. Sci. 2023, 10, 399. https://doi.org/10.3390/vetsci10060399
Kong J, Yang Y, Liu Y, Cao Y, Qiu J, Sun P, Cao X. The Pharmacokinetic and Absolute Bioavailability of Cyclosporine (Atopica for Cats®) in Cats. Veterinary Sciences. 2023; 10(6):399. https://doi.org/10.3390/vetsci10060399
Chicago/Turabian StyleKong, Jingyuan, Yuxin Yang, Yu Liu, Yuying Cao, Jicheng Qiu, Pan Sun, and Xingyuan Cao. 2023. "The Pharmacokinetic and Absolute Bioavailability of Cyclosporine (Atopica for Cats®) in Cats" Veterinary Sciences 10, no. 6: 399. https://doi.org/10.3390/vetsci10060399