

Enhancing Molecular Testing for Effective Delivery of Actionable Gene Diagnostics

Abstract
:
1. Introduction
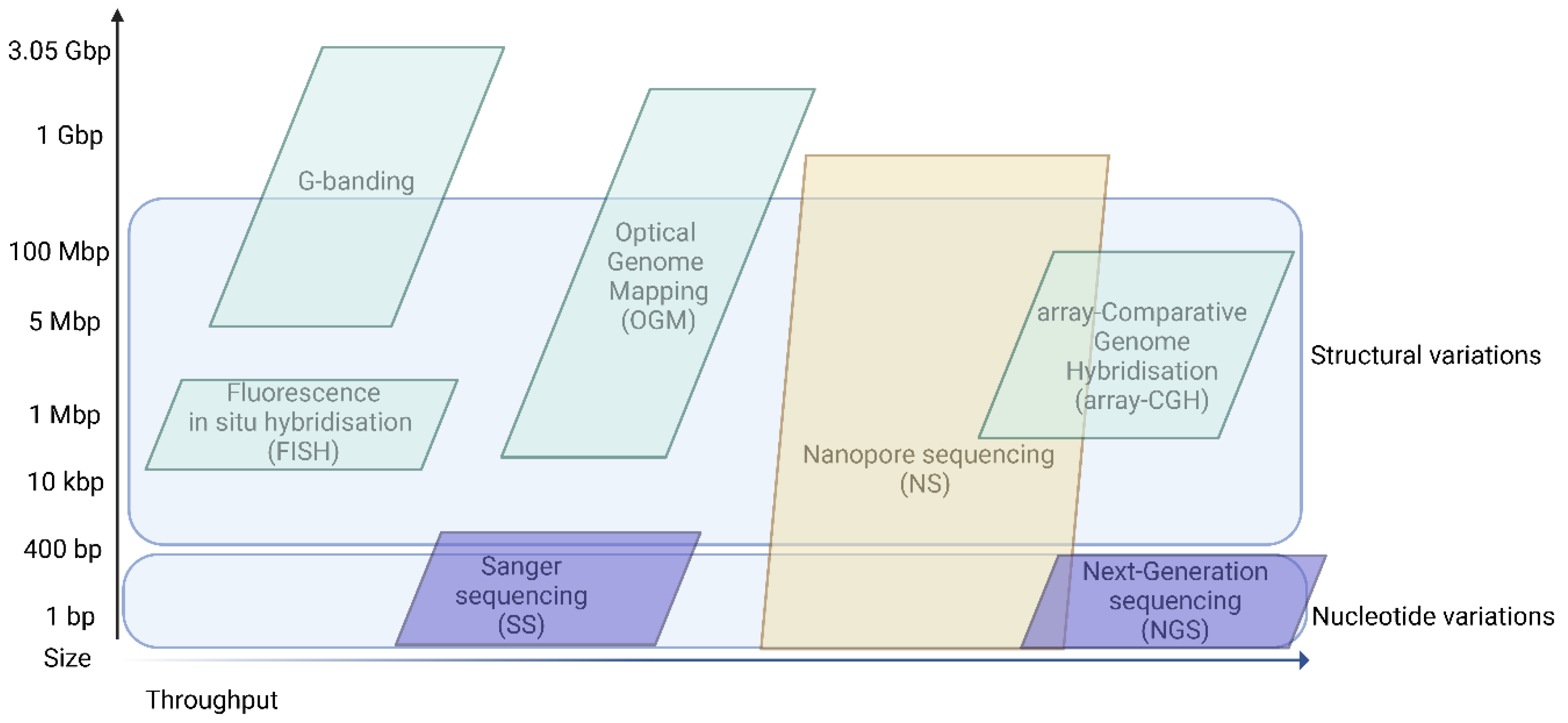
2. Molecular Testing—Timing and Approach
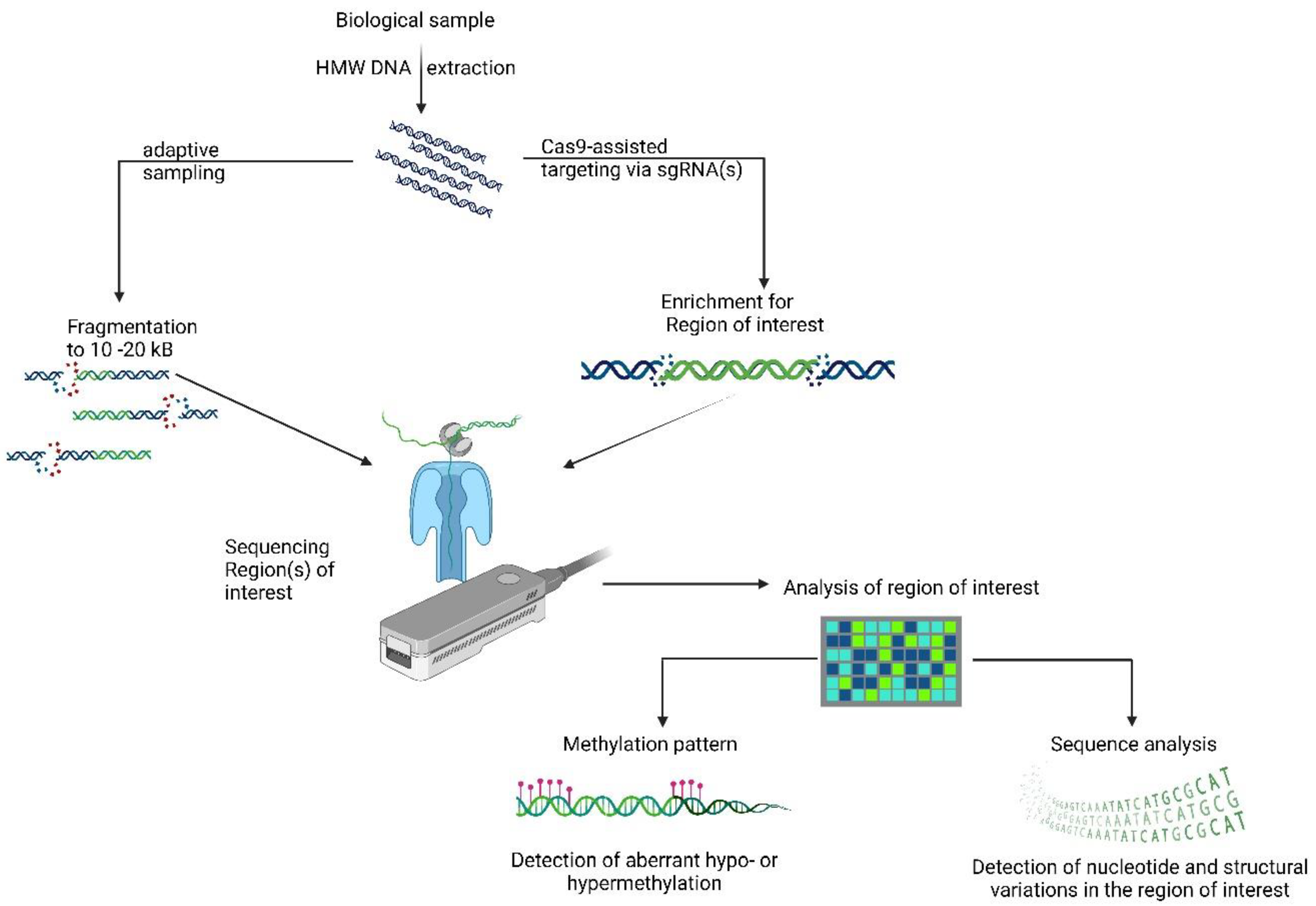
3. Nanopore Sequencing
4. Where to Fit Long-Read Sequencing in Clinical Genomics?
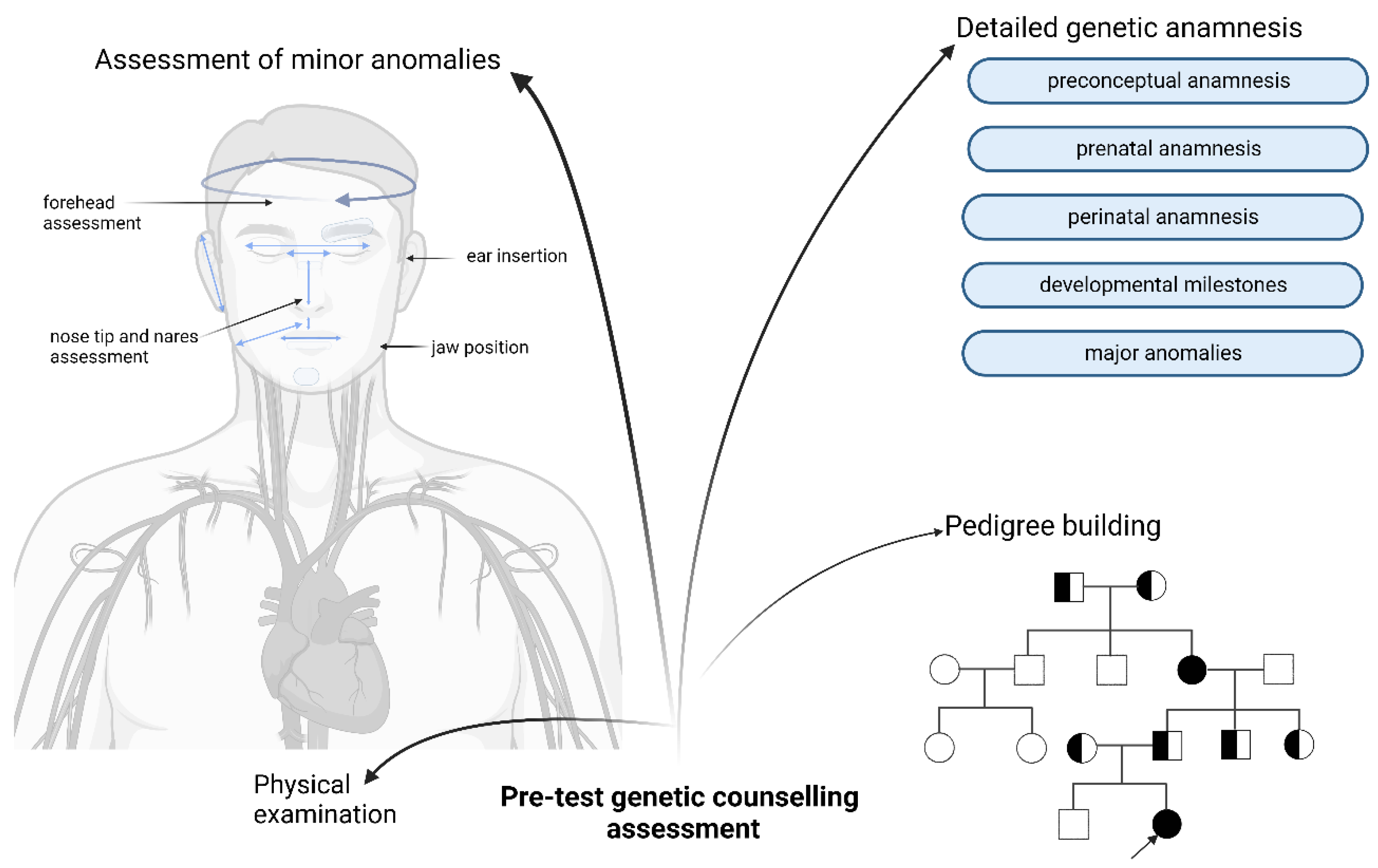
5. Genetic Counselling—State of the Art
6. Role of Pre-Test Genetic Counselling
7. Role of Post-Test Genetic Counselling
8. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics and Genomics |
| AI | artificial intelligence |
| arrayCGH | array comparative genome hybridization |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| FISH | fluorescence in situ hybridization |
| HMW DNA | high-molecular-weight DNA |
| HPO | human phenome ontology |
| nCATS | nanopore Cas9-targeted sequencing |
| MS-MLPA | methylation-sensitive multiple ligation-based assay |
| NGS | next-generation sequencing |
| NIPT | noninvasive prenatal testing |
| OGM | optical genome mapping |
| ONT | nanopore-based sequencing |
| SNV | single nucleotide variation |
| SS | Sanger sequencing |
| STR | short tandem repeats |
| TORCH | toxoplasmosis, others, rubella, cytomegalovirus and herpes simplex virus |
References
- Ferreira, C.R. The burden of rare diseases. Am. J. Med. Genet. Part A 2019, 179, 885–892. [Google Scholar] [CrossRef]
- Nguengang Wakap, S.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. Eur. J. Hum. Genet. 2020, 28, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A brief history of human disease genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Cotton, C.M.; Murray, J.C. 17—The Human Genome and Neonatal Care. In Avery’s Diseases of the Newborn, 10th ed.; Gleason, C.A., Juul, S.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 180–189.e2. Available online: https://www.sciencedirect.com/science/article/pii/B9780323401395000176 (accessed on 1 September 2022).
- Clark, M.M.; Stark, Z.; Farnaes, L.; Tan, T.Y.; White, S.M.; Dimmock, D.; Kingsmore, S.F. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med. 2018, 3, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.E.; Sulovari, A.; Wang, T.; Loucks, H.; Hoekzema, K.; Munson, K.M.; Lewis, A.P.; Fuerte, E.P.A.; Paschal, C.R.; Walsh, T.; et al. Targeted long-read sequencing identifies missing disease-causing variation. Am. J. Hum. Genet. 2021, 108, 1436–1449. [Google Scholar] [CrossRef]
- Campaña, H.; Rittler, M.; Poletta, F.A.; Gili, J.A.; Pawluk, M.S.; Scala, S.C.; Camelo, J.S.L. Minor Anomalies: Can They Predict Specific Major Defects? A Study Based on 23 Major and 14 Minor Anomalies in Over 25,000 Newborns with Birth Defects. Am. J. Perinatol. 2013, 31, 447–454. [Google Scholar] [CrossRef]
- Forrest, I.S.; Chaudhary, K.; Vy, H.M.T.; Petrazzini, B.O.; Bafna, S.; Jordan, D.M.; Rocheleau, G.; Loos, R.J.F.; Nadkarni, G.N.; Cho, J.H.; et al. Population-Based Penetrance of Deleterious Clinical Variants. JAMA 2022, 327, 350–359. [Google Scholar] [CrossRef]
- Boycott, K.M.; Hartley, T.; Adam, S.; Bernier, F.P.; Chong, K.; Fernandez, B.A.; Friedman, J.; Geraghty, M.T.; Hume, S.; Knoppers, B.; et al. The clinical application of genome-wide sequencing for monogenic diseases in Canada: Position Statement of the Canadian College of Medical Geneticists. J. Med. Genet. 2015, 52, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Haldeman-Englert, C.R.; Saitta, S.C.; Zackai, E.H. 19—The Dysmorphic Infant. In Avery’s Diseases of the Newborn, 10th ed.; Gleason, C.A., Juul, S.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 201–210.e1. Available online: https://www.sciencedirect.com/science/article/pii/B978032340139500019X (accessed on 1 September 2022).
- Dremsek, P.; Schwarz, T.; Weil, B.; Malashka, A.; Laccone, F.; Neesen, J. Optical Genome Mapping in Routine Human Genetic Diagnostics—Its Advantages and Limitations. Genes 2021, 12, 1958. [Google Scholar] [CrossRef] [PubMed]
- Gilpatrick, T.; Lee, I.; Graham, J.; Raimondeau, E.; Bowen, R.; Heron, A.; Downs, B.; Sukumar, S.; Sedlazeck, F.J.; Timp, W. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 2020, 38, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Madjunkova, S.; Sundaravadanam, Y.; Antes, R.; Abramov, R.; Chen, S.; Yin, Y.; Zuzarte, P.C.; Moskovtsev, S.I.; Jorgensen, L.G.; Baratz, A.; et al. Detection of Structural Rearrangements in Embryos. N. Engl. J. Med. 2020, 382, 2472–2474. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Liu, W.; Zhang, L.; Guo, Y.; Chen, M.; Zeng, X.; Wang, Y.; Li, Y.; Xian, J.; Du, B.; et al. Noninvasive prenatal testing for β-thalassemia by targeted nanopore sequencing combined with relative haplotype dosage (RHDO): A feasibility study. Sci. Rep. 2021, 11, 5714. [Google Scholar] [CrossRef] [PubMed]
- Gorzynski, J.E.; Goenka, S.D.; Shafin, K.; Jensen, T.D.; Fisk, D.G.; Grove, M.E.; Spiteri, E.; Pesout, T.; Monlong, J.; Baid, G.; et al. Ultrarapid Nanopore Genome Sequencing in a Critical Care Setting. N. Engl. J. Med. 2022, 386, 700–702. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Wang, J.; Bhakta, N.; Miller, V.A.; Revsine, M.; Litzow, M.R.; Paietta, E.; Fedoriw, Y.; Roberts, K.G.; Gu, Z.; Mullighan, C.G.; et al. Acute Leukemia Classification Using Transcriptional Profiles From Low-Cost Nanopore mRNA Sequencing. JCO Precis. Oncol. 2022, 6, e2100326. [Google Scholar] [CrossRef]
- Cumbo, C.; Minervini, C.F.; Orsini, P.; Anelli, L.; Zagaria, A.; Minervini, A.; Coccaro, N.; Impera, L.; Tota, G.; Parciante, E.; et al. Nanopore Targeted Sequencing for Rapid Gene Mutations Detection in Acute Myeloid Leukemia. Genes 2019, 10, 1026. [Google Scholar] [CrossRef] [Green Version]
- Karst, S.M.; Ziels, R.M.; Kirkegaard, R.H.; Sørensen, E.A.; McDonald, D.; Zhu, Q.; Knight, R.; Albertsen, M. High-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or PacBio sequencing. Nat. Methods 2021, 18, 165–169. [Google Scholar] [CrossRef]
- Ma, Z.; Li, L.; Ye, C.; Peng, M.; Zhang, Y.-P. Hybrid assembly of ultra-long Nanopore reads augmented with 10x-Genomics contigs: Demonstrated with a human genome. Genomics 2018, 111, 1896–1901. [Google Scholar] [CrossRef]
- Gabrieli, T.; Sharim, H.; Fridman, D.; Arbib, N.; Michaeli, Y.; Ebenstein, Y. Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Res. 2018, 46, e87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leija-Salazar, M.; Sedlazeck, F.J.; Toffoli, M.; Mullin, S.; Mokretar, K.; Athanasopoulou, M.; Donald, A.; Sharma, R.; Hughes, D.; Schapira, A.H.V.; et al. Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol. Genet. Genomic. Med. 2019, 7, e564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minervini, C.F.; Cumbo, C.; Orsini, P.; Brunetti, C.; Anelli, L.; Zagaria, A.; Minervini, A.; Casieri, P.; Coccaro, N.; Tota, G.; et al. TP53 gene mutation analysis in chronic lymphocytic leukemia by nanopore MinION sequencing. Diagn. Pathol. 2016, 11, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Roeck, A.; De Coster, W.; Bossaerts, L.; Cacace, R.; De Pooter, T.; Van Dongen, J.; D’Hert, S.; De Rijk, P.; Strazisar, M.; Van Broeckhoven, C.; et al. NanoSatellite: Accurate characterization of expanded tandem repeat length and sequence through whole genome long-read sequencing on PromethION. Genome Biol. 2019, 20, 229. [Google Scholar] [CrossRef] [Green Version]
- Au, C.H.; Ho, D.N.; Ip, B.B.; Wan, T.S.; Ng, M.H.; Chiu, E.K.; Chan, T.L.; Ma, E.S. Rapid detection of chromosomal translocation and precise breakpoint characterization in acute myeloid leukemia by nanopore long-read sequencing. Cancer Genet. 2019, 239, 22–25. [Google Scholar] [CrossRef] [Green Version]
- Tham, C.Y.; Tirado-Magallanes, R.; Goh, Y.; Fullwood, M.J.; Koh, B.T.; Wang, W.; Ng, C.H.; Chng, W.J.; Thiery, A.; Tenen, D.G.; et al. NanoVar: Accurate characterization of patients’ genomic structural variants using low-depth nanopore sequencing. Genome Biol. 2020, 21, 56. [Google Scholar] [CrossRef] [Green Version]
- Mariya, T.; Kato, T.; Sugimoto, T.; Miyai, S.; Inagaki, H.; Ohye, T.; Sugihara, E.; Muramatsu, Y.; Mizuno, S.; Kurahashi, H. Target enrichment long-read sequencing with adaptive sampling can determine the structure of the small supernumerary marker chromosomes. J. Hum. Genet. 2022, 67, 363–368. [Google Scholar] [CrossRef]
- Lang, K.; Surendranath, V.; Quenzel, P.; Schöfl, G.; Schmidt, A.H.; Lange, V. Full-Length HLA Class I Genotyping with the MinION Nanopore Sequencer. Methods Mol. Biol. 2018, 1802, 155–162. [Google Scholar] [CrossRef]
- Liu, C.; Xiao, F.; Hoisington-Lopez, J.; Lang, K.; Quenzel, P.; Duffy, B.; Mitra, R.D. Accurate Typing of Human Leukocyte Antigen Class I Genes by Oxford Nanopore Sequencing. J. Mol. Diagn. 2018, 20, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Duke, J.L.; Mosbruger, T.L.; Ferriola, D.; Chitnis, N.; Hu, T.; Tairis, N.; Margolis, D.J.; Monos, D.S. Resolving MiSeq-Generated Ambiguities in HLA-DPB1 Typing by Using the Oxford Nanopore Technology. J. Mol. Diagn. 2019, 21, 852–861. [Google Scholar] [CrossRef]
- Nowak, A.; Murik, O.; Mann, T.; Zeevi, D.A.; Altarescu, G. Detection of single nucleotide and copy number variants in the Fabry disease-associated GLA gene using nanopore sequencing. Sci. Rep. 2021, 11, 22372. [Google Scholar] [CrossRef] [PubMed]
- Graham, O.; Pitcher, T.; Liau, Y.; Miller, A.; Dalrymple-Alford, J.; Anderson, T.; Kennedy, M. Nanopore sequencing of the glucocerebrosidase (GBA) gene in a New Zealand Parkinson’s disease cohort. Park. Relat. Disord. 2020, 70, 36–41. [Google Scholar] [CrossRef] [Green Version]
- Lüth, T.; Laβ, J.; Schaake, S.; Wohlers, I.; Pozojevic, J.; Jamora, R.D.G.; Rosales, R.L.; Brüggemann, N.; Saranza, G.; Diesta, C.C.E.; et al. Elucidating Hexanucleotide Repeat Number and Methylation within the X-Linked Dystonia-Parkinsonism (XDP)-Related SVA Retrotransposon in TAF1 with Nanopore Sequencing. Genes 2022, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Giesselmann, P.; Brändl, B.; Raimondeau, E.; Bowen, R.; Rohrandt, C.; Tandon, R.; Kretzmer, H.; Assum, G.; Galonska, C.; Siebert, R.; et al. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat. Biotechnol. 2019, 37, 1478–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevanovski, I.; Chintalaphani, S.R.; Gamaarachchi, H.; Ferguson, J.M.; Pineda, S.S.; Scriba, C.K.; Tchan, M.; Fung, V.; Ng, K.; Cortese, A.; et al. Comprehensive genetic diagnosis of tandem repeat expansion disorders with programmable targeted nanopore sequencing. Sci. Adv. 2022, 8, eabm5386. [Google Scholar] [CrossRef]
- Bruels, C.C.; Littel, H.R.; Daugherty, A.L.; Stafki, S.; Estrella, E.A.; McGaughy, E.S.; Truong, D.; Badalamenti, J.P.; Pais, L.; Ganesh, V.S.; et al. Diagnostic capabilities of nanopore long-read sequencing in muscular dystrophy. Ann. Clin. Transl. Neurol. 2022, 9, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Lee, J.; Robinson, H.; Le, L.P.; Iafrate, A.J.; Nardi, V. A Nanopore Sequencing–Based Assay for Rapid Detection of Gene Fusions. J. Mol. Diagn. 2018, 21, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Minervini, C.F.; Cumbo, C.; Redavid, I.; Conserva, M.R.; Orsini, P.; Zagaria, A.; Anelli, L.; Coccaro, N.; Tota, G.; Impera, L.; et al. Nanopore sequencing approach for immunoglobulin gene analysis in chronic lymphocytic leukemia. Sci. Rep. 2021, 11, 17668. [Google Scholar] [CrossRef]
- Ton, K.N.T.; Cree, S.L.; Gronert-Sum, S.J.; Merriman, T.R.; Stamp, L.K.; Kennedy, M.A. Multiplexed Nanopore Sequencing of HLA-B Locus in Māori and Pacific Island Samples. Front. Genet. 2018, 9, 152. [Google Scholar] [CrossRef] [Green Version]
- Jeck, W.R.; Iafrate, A.J.; Nardi, V. Nanopore Flongle Sequencing as a Rapid, Single-Specimen Clinical Test for Fusion Detection. J. Mol. Diagn. 2021, 23, 630–636. [Google Scholar] [CrossRef]
- Martignano, F.; Munagala, U.; Crucitta, S.; Mingrino, A.; Semeraro, R.; Del Re, M.; Petrini, I.; Magi, A.; Conticello, S.G. Nanopore sequencing from liquid biopsy: Analysis of copy number variations from cell-free DNA of lung cancer patients. Mol. Cancer 2021, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chen, Y.; Shen, C.; Li, L.; Li, Q.; Tan, K.; Huang, H.; Hu, G. Breakpoint mapping of a t(9;22;12) chronic myeloid leukaemia patient with e14a3 BCR-ABL1 transcript using Nanopore sequencing. J. Gene Med. 2020, 23, e3276. [Google Scholar] [CrossRef] [PubMed]
- Cumbo, C.; Orsini, P.; Anelli, L.; Zagaria, A.; Minervini, C.F.; Coccaro, N.; Tota, G.; Impera, L.; Parciante, E.; Conserva, M.R.; et al. Nanopore sequencing sheds a light on the FLT3 gene mutations complexity in acute promyelocytic leukemia. Leuk. Lymphoma 2020, 62, 1219–1225. [Google Scholar] [CrossRef]
- Zascavage, R.R.; Thorson, K.; Planz, J.V. Nanopore sequencing: An enrichment-free alternative to mitochondrial DNA sequencing. ELECTROPHORESIS 2018, 40, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.T.; Lee, K.; Abul-Husn, N.S.; Amendola, L.M.; Brothers, K.; Chung, W.K.; Gollob, M.H.; Gordon, A.S.; Harrison, S.M.; Hershberger, R.E.; et al. ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. Off. J. Am. Coll. Med. Genet. 2022, 24, 1407–1414. [Google Scholar] [CrossRef]
- Schneider, H.; Faschingbauer, F.; Schuepbach-Mallepell, S.; Körber, I.; Wohlfart, S.; Dick, A.; Wahlbuhl, M.; Kowalczyk-Quintas, C.; Vigolo, M.; Kirby, N.; et al. Prenatal Correction of X-Linked Hypohidrotic Ectodermal Dysplasia. N. Engl. J. Med. 2018, 378, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Peranteau, W.H.; Flake, A.W. The Future of In Utero Gene Therapy. Mol. Diagn. Ther. 2020, 24, 135–142. [Google Scholar] [CrossRef]
- Patch, C.; Middleton, A. Genetic counselling in the era of genomic medicine. Br. Med. Bull. 2018, 126, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Aoi, H.; Mizuguchi, T.; Suzuki, T.; Makino, S.; Yamamoto, Y.; Takeda, J.; Maruyama, Y.; Seyama, R.; Takeuchi, S.; Uchiyama, Y.; et al. Whole exome sequencing of fetal structural anomalies detected by ultrasonography. J. Hum. Genet. 2021, 66, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Founti, P.; Topouzis, F.; Van Koolwijk, L.; Traverso, C.E.; Pfeiffer, N.; Viswanathan, A.C. Biobanks and the importance of detailed phenotyping: A case study--the European Glaucoma Society GlaucoGENE project. Br. J. Ophthalmol. 2009, 93, 577–581. [Google Scholar] [CrossRef]
- Corbin, L.J.; Tan, V.Y.; Hughes, D.A.; Wade, K.H.; Paul, D.S.; Tansey, K.E.; Butcher, F.; Dudbridge, F.; Howson, J.M.; Jallow, M.W.; et al. Formalising recall by genotype as an efficient approach to detailed phenotyping and causal inference. Nat. Commun. 2018, 9, 711. [Google Scholar] [CrossRef] [Green Version]
- Maddirevula, S.; Alsahli, S.; Alhabeeb, L.; Patel, N.; Alzahrani, F.; Shamseldin, H.E.; Anazi, S.; Ewida, N.; Alsaif, H.S.; Mohamed, J.Y.; et al. Expanding the phenome and variome of skeletal dysplasia. Genet. Med. 2018, 20, 1609–1616. [Google Scholar] [CrossRef]
- Hennekam, R.C.; Biesecker, L.G. Next-generation sequencing demands next-generation phenotyping. Hum. Mutat. 2012, 33, 884–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snoek, R.; van Eerde, A.M.; Knoers, N.V.A.M. Importance of reliable variant calling and clear phenotyping when reporting on gene panel testing in renal disease. Kidney Int. 2017, 92, 1325–1327. [Google Scholar] [CrossRef]
- Allanson, J.E.; Cunniff, C.; Hoyme, H.; McGaughran, J.; Muenke, M.; Neri, G. Elements of morphology: Standard terminology for the head and face. Am. J. Med. Genet. Part A 2009, 149A, 6–28. [Google Scholar] [CrossRef] [Green Version]
- Gripp, K.W.; Slavotinek, A.M.; Hall, J.G.; Allanson, J.E. Handbook of Physical Measurements; Oxford University Press: Oxford, UK, 2013. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Adam, M.P.; Chung, B.H.; Kosaki, K.; Menke, L.A.; White, S.M.; Carey, J.C.; Hennekam, R.C.M. Elements of morphology: Standard terminology for the trunk and limbs. Am. J. Med. Genet. Part A 2022, 188, 3191–3228. [Google Scholar] [CrossRef]
- Marwaha, A.; Chitayat, D.; Meyn, M.S.; Mendoza-Londono, R.; Chad, L. The point-of-care use of a facial phenotyping tool in the genetics clinic: Enhancing diagnosis and education with machine learning. Am. J. Med. Genet. Part A 2021, 185, 1151–1158. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Williams, M.S.; Basma, N.J.; Amaral, F.M.R.; Williams, G.; Weightman, J.P.; Breitwieser, W.; Nelson, L.; Taylor, S.S.; Wiseman, D.H.; Somervaille, T.C.P. Targeted nanopore sequencing for the identification of ABCB1 promoter translocations in cancer. BMC Cancer 2020, 20, 1075. [Google Scholar] [CrossRef]
- Kuderna, L.F.K.; Solís-Moruno, M.; Batlle-Masó, L.; Julià, E.; Lizano, E.; Anglada, R.; Ramírez, E.; Bote, A.; Tormo, M.; Marquès-Bonet, T.; et al. Flow Sorting Enrichment and Nanopore Sequencing of Chromosome 1 From a Chinese Individual. Front. Genet. 2020, 10, 1315. [Google Scholar] [CrossRef] [Green Version]
- Karousis, E.D.; Gypas, F.; Zavolan, M.; Mühlemann, O. Nanopore sequencing reveals endogenous NMD-targeted isoforms in human cells. Genome Biol. 2021, 22, 223. [Google Scholar] [CrossRef]
- Lee, I.; Razaghi, R.; Gilpatrick, T.; Molnar, M.; Gershman, A.; Sadowski, N.; Sedlazeck, F.J.; Hansen, K.D.; Simpson, J.T.; Timp, W. Simultaneous profiling of chromatin accessibility and methylation on human cell lines with nanopore sequencing. Nat. Methods 2020, 17, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region of Interest | Sample | DNA/RNA Extraction | Library Prep | Sequencing Platform | Results | Validation | Reference |
|---|---|---|---|---|---|---|---|
| Genetic regions of interest for inborn errors of metabolism | |||||||
| GLA | Blood | Flexigene DNA kit (Qiagen) | PBK004 | MinION | Detected GLA nucleotide variations | Known GLA variants were sequenced | [33] |
| GBA | Blood | Phenol-chloroform salting out | LSK-109 | GridION | Detected variants, SNV | SS | [34] |
| PAH | Blood, saliva, fibroblasts | NA | LSK-109 | GridION | Successful SV, complex rearrangements, SNV detection with adaptive sampling | SS, Southern blot | [8] |
| Genetic regions of interest for intellectual disability | |||||||
| TAF1 | Blood | DNA Midi kit (Qiagen) | LSK-109 | GridION | Detected repeat expansions | Fluorescence-based PCR | [35] |
| C9orf72, FMR1 | hiPSC from patients | Phenol-chloroform extractions | LSK-108 or LSK-109 | MinION | STR | Southern blot | [36] |
| RFC1, NOTCH2NLC, FXN, AR, DMPK | Blood | Qiagen Gentra PureGene blood kit (NSW) or QIAsymphony DSP DNA Midi Kit | LSK109 or LSK110 | MinION or GridION | STR and methylation profiling | RP-PCR and Southern blot | [37] |
| Genetic regions of interest for skeletal and or heart disorders | |||||||
| DMD | Blood and saliva | DNA extraction | LSK-109 or LSK-110 | MinION or GridION | Detected SVs and SNVs | SS | [38] |
| ALMS1, DMD, ABCA4, AGL, XYLT1 and other ROI | Blood, saliva, fibroblasts | NA | LSK-109 | GridION | Successful SV, complex rearrangements, SNV detection with adaptive sampling | SS, Southern blot | [8] |
| Genetic regions of interest tumour predisposition | |||||||
| PML-RARA | Blood and bone marrow | Promega Maxwell Instrument | LSK-108 | MinION | Gene fusion detection | NGS—Illumina | [39] |
| IGHV | Blood | QIAamp DNA blood mini kit | LSK-109 | MinION | Detection of IGHV small subclones | SS | [40] |
| BRCA1, KRT19, BRAF, KRAS, TP53 | Breast tumor | MasterPure kit (Lucigen) | LSK-109 | MinION | SNV, SV, methylation by Cas9-approach | Illumina WGBS | [14] |
| HLAB2 | Blood | Guanidium–HCl based chloroform extraction | NSK007 | MinION | HLA-B genotyping | SS | [41] |
| BCR-ABL1, FGFR2 fusions, | Hematologic and solid tissue specimens | RNA extraction Promega | LSK-108 | MinION, Flongle | Gene fusion detection | NGS | [42] |
| EGFR | Blood plasma | QIAamp Circulating Nucleic Acid Kit | LSK-109 | MinION | EGFR amplifications | NGS, SS | [43] |
| NPM1, FLT3, CEBPA, TP53, IDH1 and IDH2 | Bone marrow | QIAamp DNA Blood mini kit | LSK-108 | MinION | SNV, indel detection | SS | [20] |
| BCR-ABL1 | Blood | Blood genomic DNA mini kit | LSK-108 | MinION | Specific positions of translocation | SS | [44] |
| FLT3 | Blood | NA | LSK-108 | MinION | FLT3 allelic mutations | NGS—Ion Torrent | [45] |
| mtDNA | |||||||
| mtDNA | Blood | QIAamp Mini Blood Kit | NSK007 or RAD001 | MinION | SNV, homopolymer, dinucleotide repeat | NIST-traceable mtDNA sequencing standard | [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, Á.F.; Némethi, Z.; Abonyi, T.; Fekete, G.; Kovács, G.T. Enhancing Molecular Testing for Effective Delivery of Actionable Gene Diagnostics. Bioengineering 2022, 9, 745. https://doi.org/10.3390/bioengineering9120745
Kovács ÁF, Némethi Z, Abonyi T, Fekete G, Kovács GT. Enhancing Molecular Testing for Effective Delivery of Actionable Gene Diagnostics. Bioengineering. 2022; 9(12):745. https://doi.org/10.3390/bioengineering9120745
Chicago/Turabian StyleKovács, Árpád Ferenc, Zaránd Némethi, Tünde Abonyi, György Fekete, and Gábor T. Kovács. 2022. "Enhancing Molecular Testing for Effective Delivery of Actionable Gene Diagnostics" Bioengineering 9, no. 12: 745. https://doi.org/10.3390/bioengineering9120745