
scGENA: A Single-Cell Gene Coexpression Network Analysis Framework for Clustering Cell Types and Revealing Biological Mechanisms



Abstract
:
1. Introduction
2. Materials and Methods
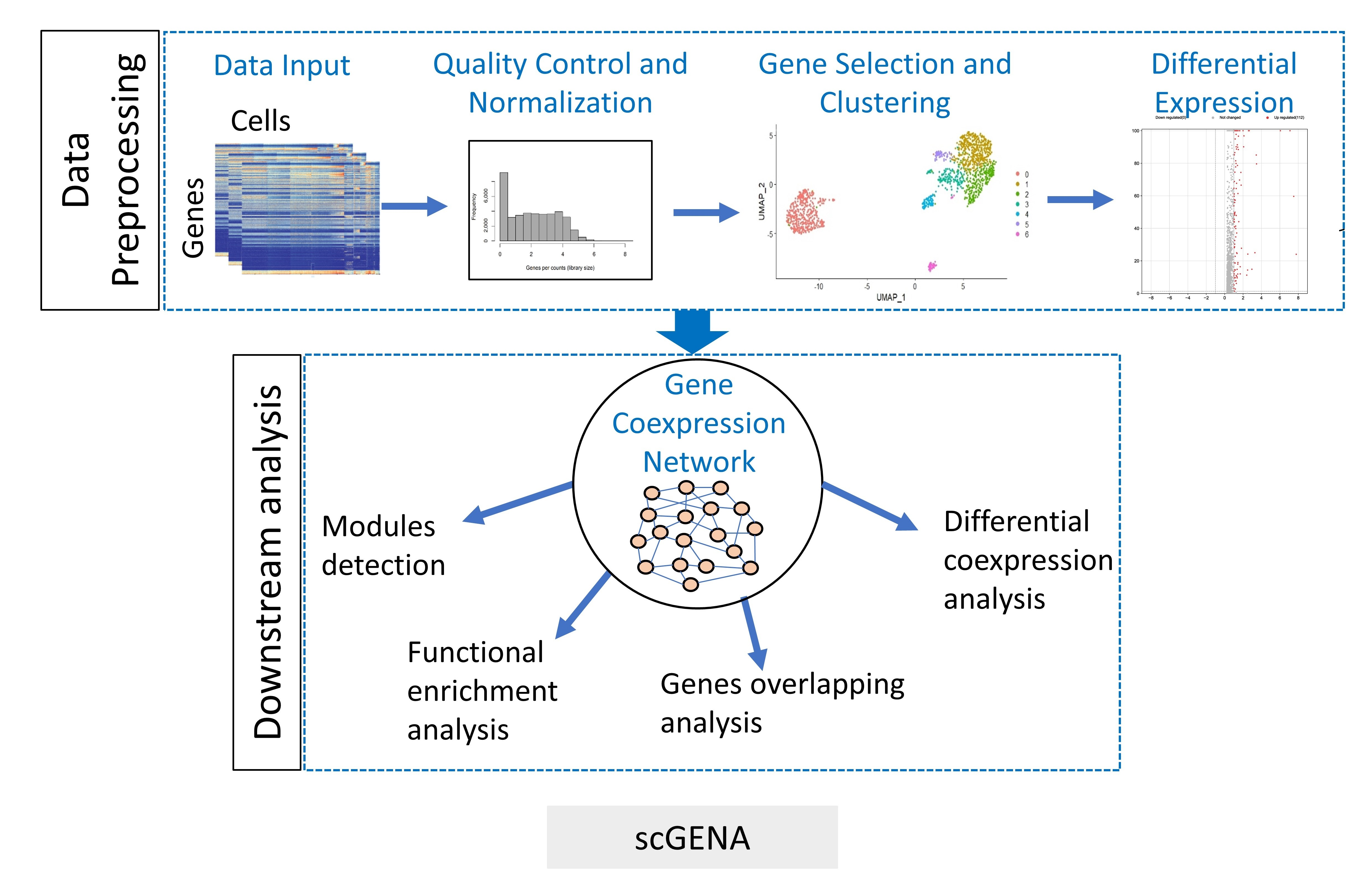
2.1. Overview of scGENA
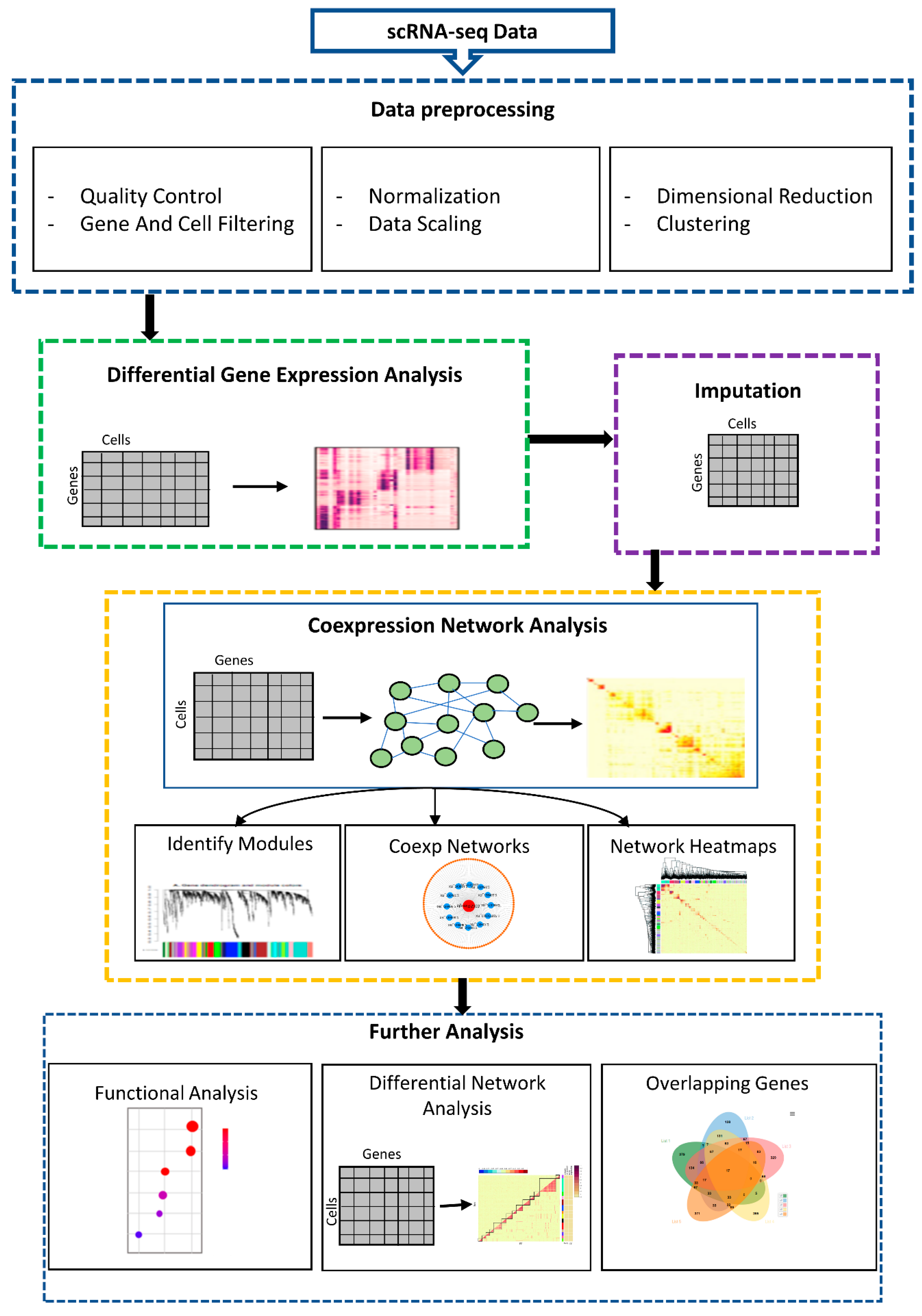
2.2. Preprocessing of scRNA-seq
2.3. Differential Expression (DE) Analysis
2.4. Data Imputation
2.5. Gene Coexpression Networks (GCNs) Analysis
3. Results and Discussion
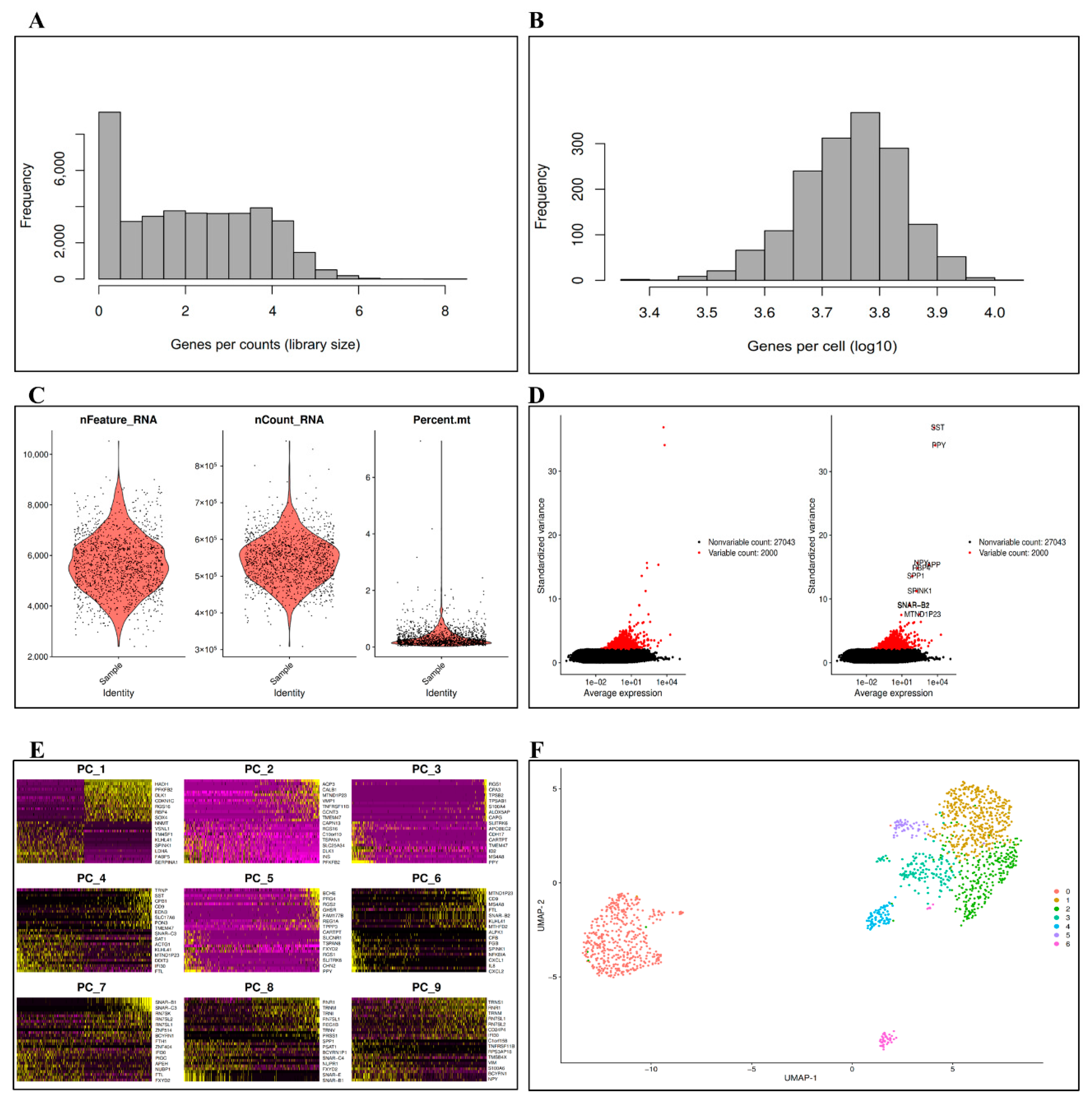
3.1. Data Preprocessing
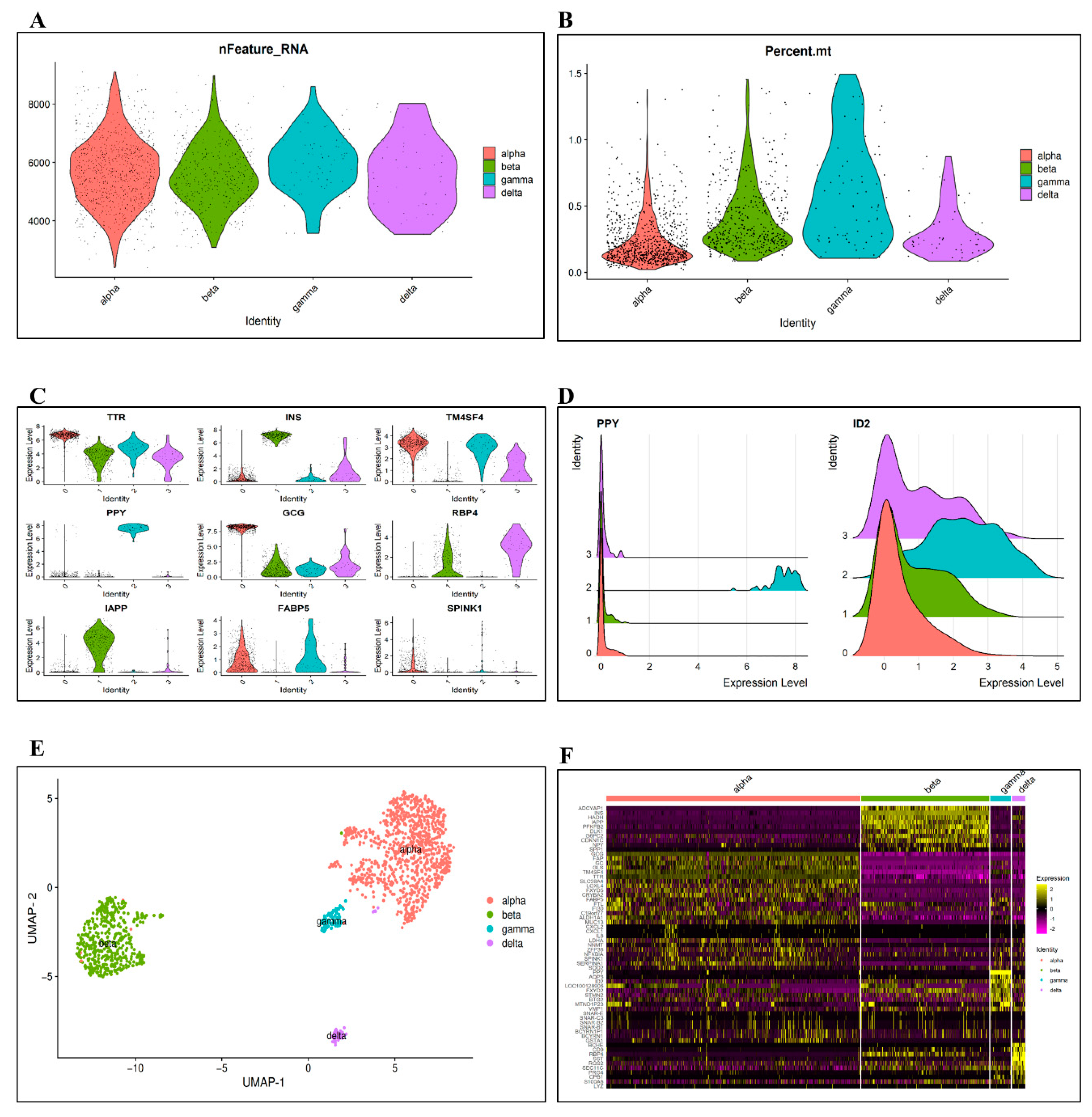
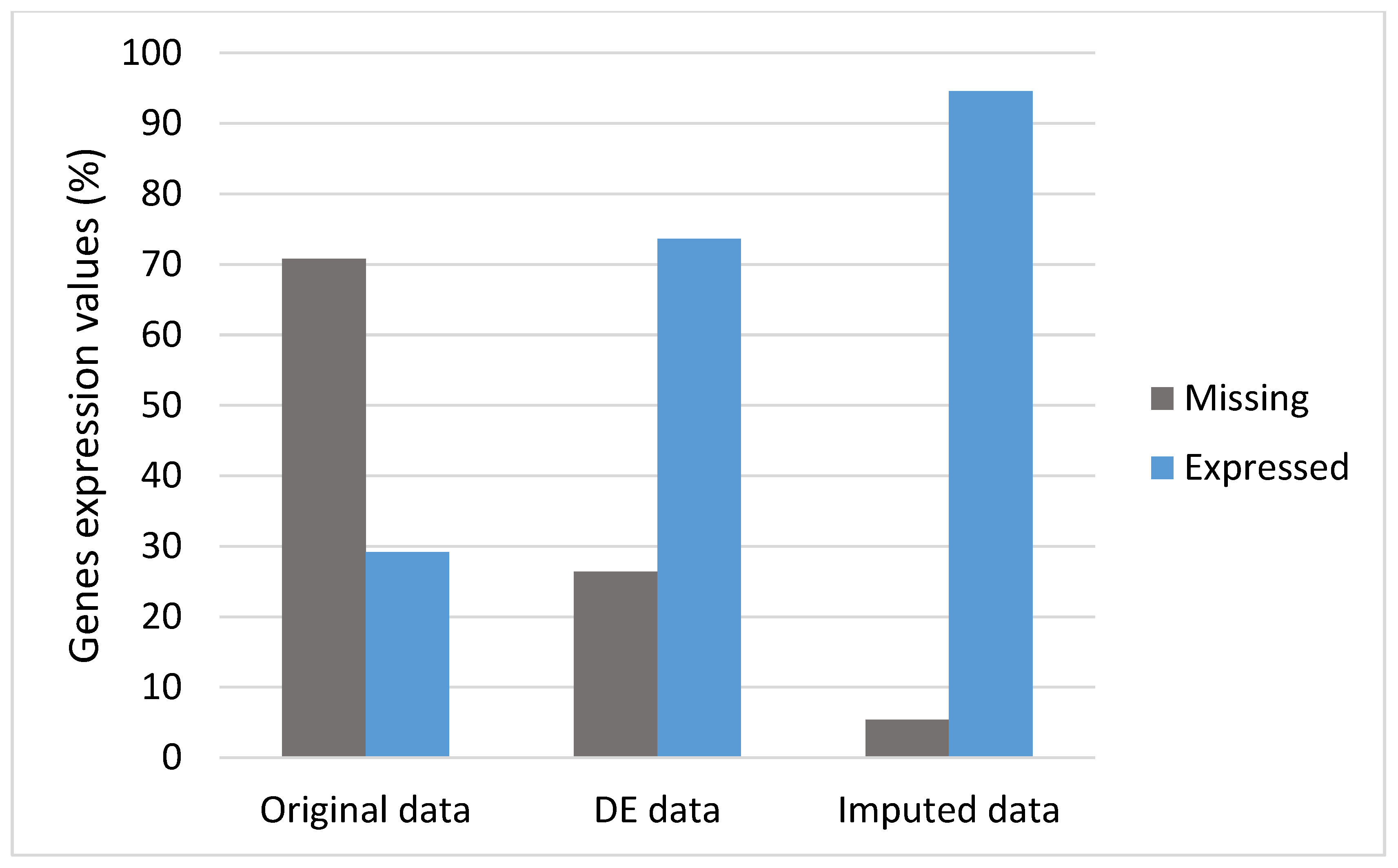
3.2. DEs and Imputation
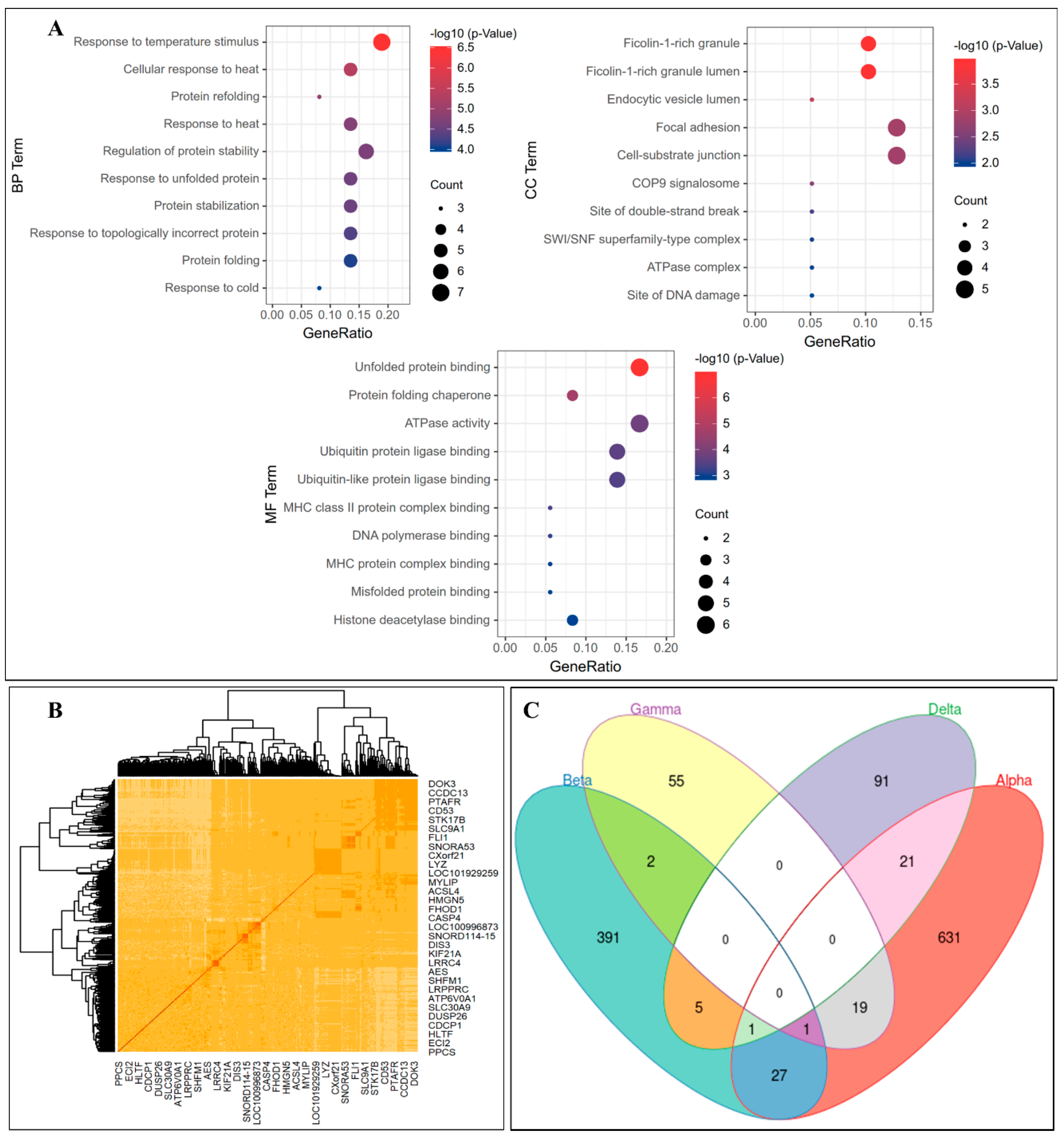
3.3. Gene Coexpression Networks Analysis
3.4. Further Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hwang, B.; Lee, J.H.; Bang, D. Single-Cell RNA Sequencing Technologies and Bioinformatics Pipelines. Exp. Mol. Med. 2018, 50, 96. [Google Scholar] [CrossRef] [Green Version]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA Sequencing: The Teenage Years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative Isoform Regulation in Human Tissue Transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, A.; Beaudin, A.E.; Olsen, H.E.; Jain, M.; Cole, C.; Palmer, T.; DuBois, R.M.; Forsberg, E.C.; Akeson, M.; Vollmers, C. Nanopore Long-Read RNAseq Reveals Widespread Transcriptional Variation among the Surface Receptors of Individual B Cells. Nat. Commun. 2017, 8, 16027. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, A.; Rookmaaker, M.B.; Joles, J.A.; Kramann, R.; Nguyen, T.Q.; van Griensven, M.; LaPointe, V.L.S. Defining the Variety of Cell Types in Developing and Adult Human Kidneys by Single-Cell RNA Sequencing. NPJ Regen. Med. 2021, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A. MRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Eberwine, J.; Sul, J.-Y.; Bartfai, T.; Kim, J. The Promise of Single-Cell Sequencing. Nat. Methods 2014, 11, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Suvà, M.L. Deciphering Human Tumor Biology by Single-Cell Expression Profiling. Annu. Rev. Cancer Biol. 2019, 3, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Saelens, W.; Cannoodt, R.; Todorov, H.; Saeys, Y. A Comparison of Single-Cell Trajectory Inference Methods. Nat. Biotechnol. 2019, 37, 547–554. [Google Scholar] [CrossRef]
- Wang, T.; Nabavi, S. SigEMD: A Powerful Method for Differential Gene Expression Analysis in Single-Cell RNA Sequencing Data. Methods 2018, 145, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic Gene Expression in a Single Cell. Science 2002, 297, 1183–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komili, S.; Silver, P.A. Coupling and Coordination in Gene Expression Processes: A Systems Biology View. Nat. Rev. Genet. 2008, 9, 38–48. [Google Scholar] [CrossRef]
- Furlong, L.I. Human Diseases through the Lens of Network Biology. Trends Genet. 2013, 29, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Gysi, D.M.; de Fragoso, T.M.; Zebardast, F.; Bertoli, W.; Busskamp, V.; Almaas, E.; Nowick, K. Whole Transcriptomic Network Analysis Using Co-Expression Differential Network Analysis (CoDiNA). PLoS ONE 2020, 15, e0240523. [Google Scholar] [CrossRef]
- Wang, J.; Xia, S.; Arand, B.; Zhu, H.; Machiraju, R.; Huang, K.; Ji, H.; Qian, J. Single-Cell Co-Expression Analysis Reveals Distinct Functional Modules, Co-Regulation Mechanisms and Clinical Outcomes. PLoS Comput. Biol. 2016, 12, e1004892. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hu, L.; Wang, Y.; Sun, W.; Yang, C. Single Cell Gene Co-Expression Network Reveals FECH/CROT Signature as a Prognostic Marker. Cells 2019, 8, 698. [Google Scholar] [CrossRef] [Green Version]
- Elo, L.L.; Järvenpää, H.; Orešič, M.; Lahesmaa, R.; Aittokallio, T. Systematic Construction of Gene Coexpression Networks with Applications to Human T Helper Cell Differentiation Process. Bioinformatics 2007, 23, 2096–2103. [Google Scholar] [CrossRef] [Green Version]
- Reverter, A.; Chan, E.K.F. Combining Partial Correlation and an Information Theory Approach to the Reversed Engineering of Gene Co-Expression Networks. Bioinformatics 2008, 24, 2491–2497. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.W.; Beech, D.J.; Wheatcroft, S.B. Advantages of CEMiTool for Gene Co-Expression Analysis of RNA-Seq Data. Comput. Biol. Med. 2020, 125, 103975. [Google Scholar] [CrossRef]
- Rexach, J.E.; Polioudakis, D.; Yin, A.; Swarup, V.; Chang, T.S.; Nguyen, T.; Sarkar, A.; Chen, L.; Huang, J.; Lin, L.-C.; et al. Tau Pathology Drives Dementia Risk-Associated Gene Networks toward Chronic Inflammatory States and Immunosuppression. Cell Rep. 2020, 33, 108398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer’s Disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Wang, J.; Torre, E.; Dueck, H.; Shaffer, S.; Bonasio, R.; Murray, J.I.; Raj, A.; Li, M.; Zhang, N.R. SAVER: Gene Expression Recovery for Single-Cell RNA Sequencing. Nat. Methods 2018, 15, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Li, W.V.; Li, Y. ScLink: Inferring Sparse Gene Co-Expression Networks from Single-Cell Expression Data. Genom. Proteom. Bioinform. 2021, 19, 475–492. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Kim, J.; Okamoto, H.; Ni, M.; Wei, Y.; Adler, C.; Murphy, A.J.; Yancopoulos, G.D.; Lin, C.; Gromada, J. RNA Sequencing of Single Human Islet Cells Reveals Type 2 Diabetes Genes. Cell Metab. 2016, 24, 608–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soneson, C.; Robinson, M.D. Bias, Robustness and Scalability in Single-Cell Differential Expression Analysis. Nat. Methods 2018, 15, 255–261. [Google Scholar] [CrossRef]
- Finak, G.; McDavid, A.; Yajima, M.; Deng, J.; Gersuk, V.; Shalek, A.K.; Slichter, C.K.; Miller, H.W.; McElrath, M.J.; Prlic, M.; et al. MAST: A Flexible Statistical Framework for Assessing Transcriptional Changes and Characterizing Heterogeneity in Single-Cell RNA Sequencing Data. Genome Biol. 2015, 16, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDavid, A.; Finak, G.; Yajima, M. MAST: Model-Based Analysis of Single Cell Transcriptomics. R Package Version 1.22.0. 2022. Available online: https://github.com/RGLab/MAST/ (accessed on 1 September 2020).
- Li, X.; Liu, L.; Goodall, G.J.; Schreiber, A.; Xu, T.; Li, J.; Le, T.D. A Novel Single-Cell Based Method for Breast Cancer Prognosis. PLoS Comput. Biol. 2020, 16, e1008133. [Google Scholar] [CrossRef]
- van Dijk, D.; Sharma, R.; Nainys, J.; Yim, K.; Kathail, P.; Carr, A.J.; Burdziak, C.; Moon, K.R.; Chaffer, C.L.; Pattabiraman, D.; et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 2018, 174, 716–729.e27. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, S. Comparison of Computational Methods for Imputing Single-Cell RNA-Sequencing Data. IEEE/ACM Trans. Comput. Biol. Bioinf. 2017, 17, 376–389. [Google Scholar] [CrossRef]
- Iacono, G.; Massoni-Badosa, R.; Heyn, H. Single-Cell Transcriptomics Unveils Gene Regulatory Network Plasticity. Genome Biol. 2019, 20, 110. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.-P. Quantifying Gene Regulatory Relationships with Association Measures: A Comparative Study. Front. Genet. 2017, 8, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mc Mahon, S.S.; Lenive, O.; Filippi, S.; Stumpf, M.P.H. Information Processing by Simple Molecular Motifs and Susceptibility to Noise. J. R. Soc. Interface 2015, 12, 20150597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, P.E.; Lafitte, F.; Bontempi, G. Minet: A R/Bioconductor Package for Inferring Large Transcriptional Networks Using Mutual Information. BMC Bioinform. 2008, 9, 461. [Google Scholar] [CrossRef] [PubMed]
- Lytal, N.; Ran, D.; An, L. Normalization Methods on Single-Cell RNA-Seq Data: An Empirical Survey. Front. Genet. 2020, 11, 41. [Google Scholar] [CrossRef]
- Kiselev, V.Y.; Andrews, T.S.; Hemberg, M. Challenges in Unsupervised Clustering of Single-Cell RNA-Seq Data. Nat. Rev. Genet. 2019, 20, 273–282. [Google Scholar] [CrossRef]
- Abdi, H.; Williams, L.J. Principal Component Analysis. WIREs Comput. Stat. 2010, 2, 433–459. [Google Scholar] [CrossRef]
- Duò, A.; Robinson, M.D.; Soneson, C. A Systematic Performance Evaluation of Clustering Methods for Single-Cell RNA-Seq Data. F1000 Research 2020, 7, 1141. [Google Scholar] [CrossRef]
- Luecken, M.D.; Theis, F.J. Current Best Practices in Single-Cell RNA-Seq Analysis: A Tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef]
- Vieth, B.; Ziegenhain, C.; Parekh, S.; Enard, W.; Hellmann, I. PowsimR: Power Analysis for Bulk and Single Cell RNA-Seq Experiments. Bioinformatics 2017, 33, 3486–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dam, S.; Võsa, U.; van der Graaf, A.; Franke, L.; de Magalhães, J.P. Gene Co-Expression Analysis for Functional Classification and Gene–Disease Predictions. Brief. Bioinform. 2017, 19, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Yan, X.; Liang, Z.; Zheng, R.; Li, M. MKG: A Mutual Information Based Method to Infer Single Cell Gene Regulatory Network. In Proceedings of the 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Houston, TX, USA, 9–12 December 2021; pp. 223–228. [Google Scholar]
- WGCNA: R Package for Performing Weighted Gene Co-Expression Network Analysis. Available online: https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/ (accessed on 18 July 2022).
- Morabito, S.; Miyoshi, E.; Michael, N.; Swarup, V. Integrative Genomics Approach Identifies Conserved Transcriptomic Networks in Alzheimer’s Disease. Hum. Mol. Genet. 2020, 29, 2899–2919. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. Protein Misfolding in the Endoplasmic Reticulum as a Conduit to Human Disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liang, Z.; Xiang, X.; Liu, L.; Yang, H.; Tang, G. Identification and Validation of Hub Genes in Acute Pancreatitis and Hypertriglyceridemia. Diabetes Metab. Syndr. Obes. 2022, 15, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Yong, J.; Johnson, J.D.; Arvan, P.; Han, J.; Kaufman, R.J. Therapeutic Opportunities for Pancreatic β-Cell ER Stress in Diabetes Mellitus. Nat. Rev. Endocrinol. 2021, 17, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Brown, J.B.; Orsini, L.; Pan, Z.; Hu, G.; He, S. MODA: MODA: MOdule Differential Analysis for Weighted Gene Co-Expression Network. R Package Version 1.22.0. 2022. Available online: https://doi.org/10.48550/arXiv.1605.04739 (accessed on 1 September 2020). [CrossRef]
- Khan, A.; Mathelier, A. Intervene: A Tool for Intersection and Visualization of Multiple Gene or Genomic Region Sets. BMC Bioinform. 2017, 18, 287. [Google Scholar] [CrossRef] [Green Version]
- Moede, T.; Leibiger, I.B.; Berggren, P.-O. Alpha Cell Regulation of Beta Cell Function. Diabetologia 2020, 63, 2064–2075. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO No. | Type of Cells | Cells | Features | Organism | Protocol | Ref. |
|---|---|---|---|---|---|---|
| GSE81608 | α- β- δ- PP | 886 472 49 85 | 39,851 | Homo sapiens | SMARTer | Xin et al., 2016 [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Algabri, Y.A.; Li, L.; Liu, Z.-P. scGENA: A Single-Cell Gene Coexpression Network Analysis Framework for Clustering Cell Types and Revealing Biological Mechanisms. Bioengineering 2022, 9, 353. https://doi.org/10.3390/bioengineering9080353
Algabri YA, Li L, Liu Z-P. scGENA: A Single-Cell Gene Coexpression Network Analysis Framework for Clustering Cell Types and Revealing Biological Mechanisms. Bioengineering. 2022; 9(8):353. https://doi.org/10.3390/bioengineering9080353
Chicago/Turabian StyleAlgabri, Yousif A., Lingyu Li, and Zhi-Ping Liu. 2022. "scGENA: A Single-Cell Gene Coexpression Network Analysis Framework for Clustering Cell Types and Revealing Biological Mechanisms" Bioengineering 9, no. 8: 353. https://doi.org/10.3390/bioengineering9080353