

Quercetin-Induced Glutathione Depletion Sensitizes Colorectal Cancer Cells to Oxaliplatin

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Lines and Culture Conditions
2.3. Cell Viability Assay
2.4. GR Activity Assay
2.5. GSH Level Assay
2.6. Measurements of Intracellular ROS
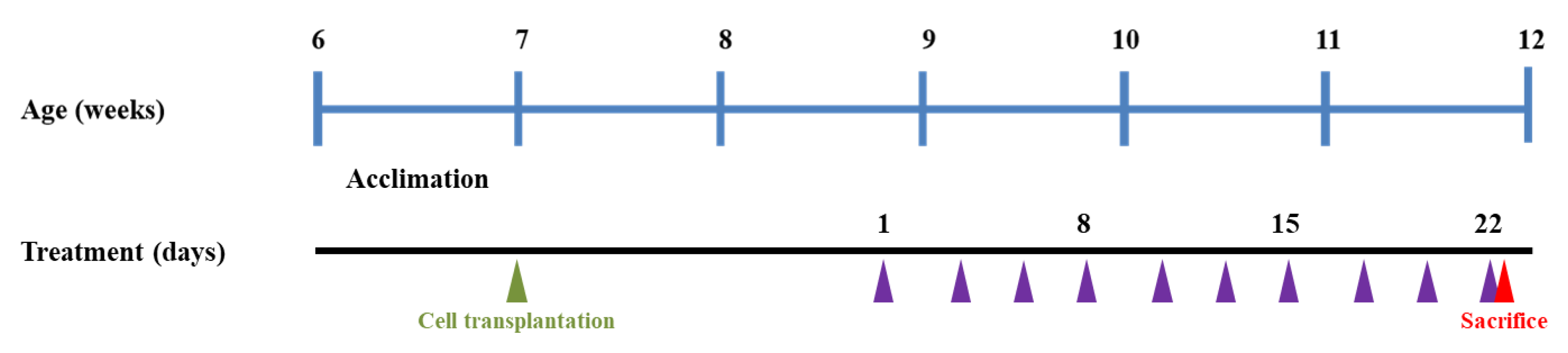
2.7. Animal Experimental Design
2.8. Measurement of Body Weight and Tumor Growth
2.9. Western Blotting
2.10. Reverse-Transcription-Quantitative PCR (RT-qPCR)
2.11. Statistical Analysis
3. Results
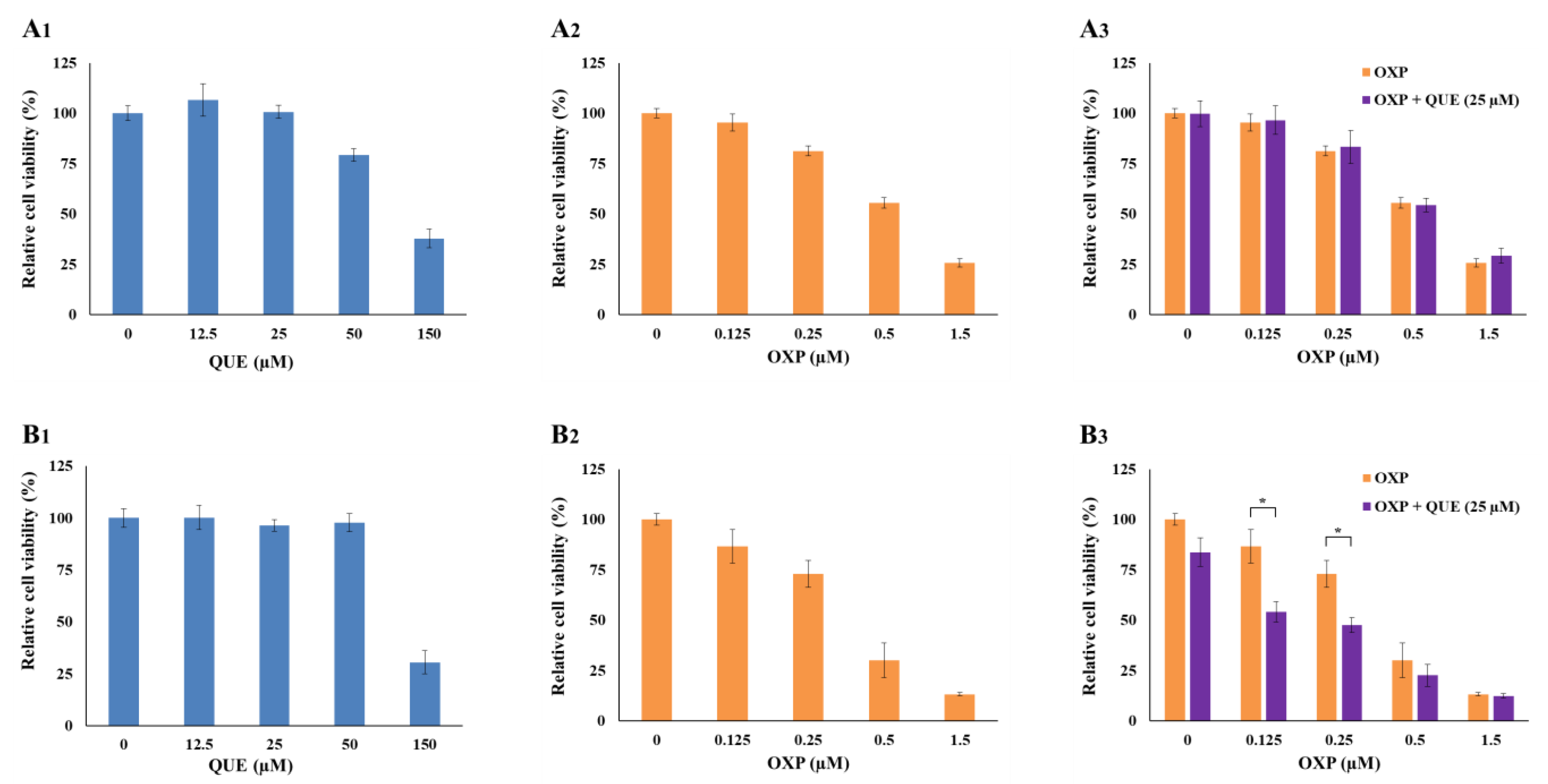
3.1. QUE Enhanced OXP-Mediated Suppression of HCT116 Cell Proliferation
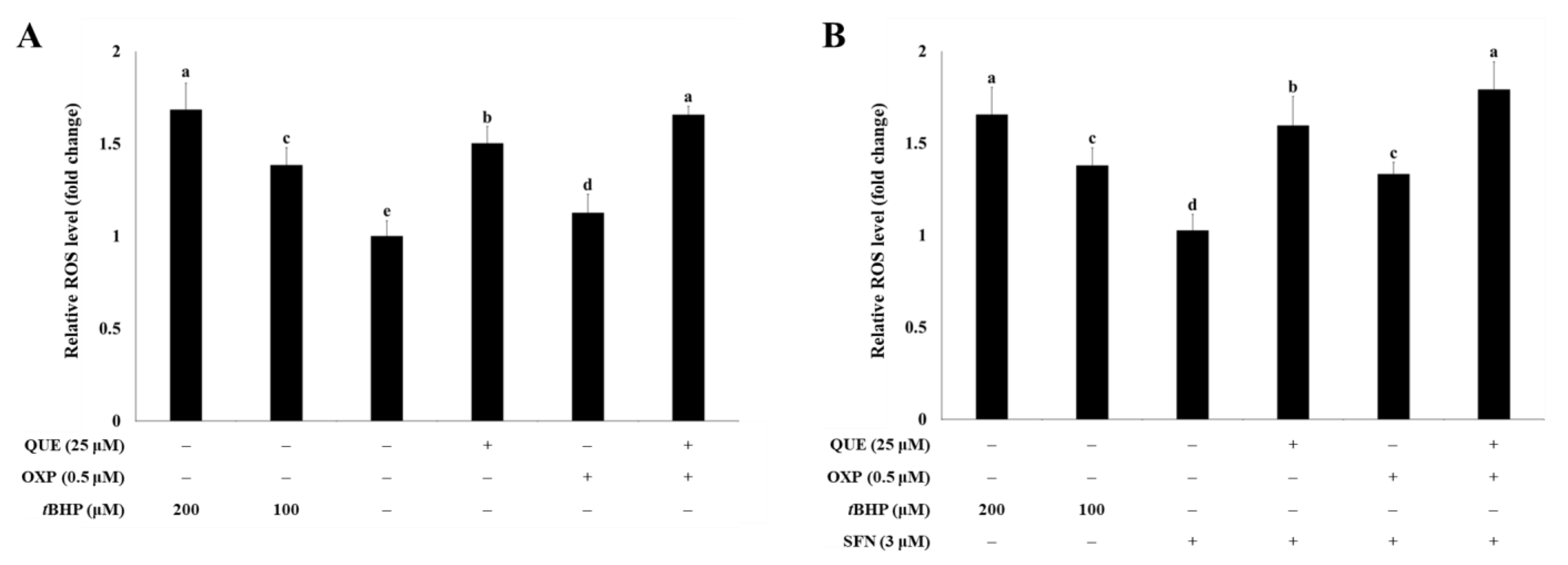
3.2. QUE Enhanced OXP-Induced ROS Production in HCT116 Cells
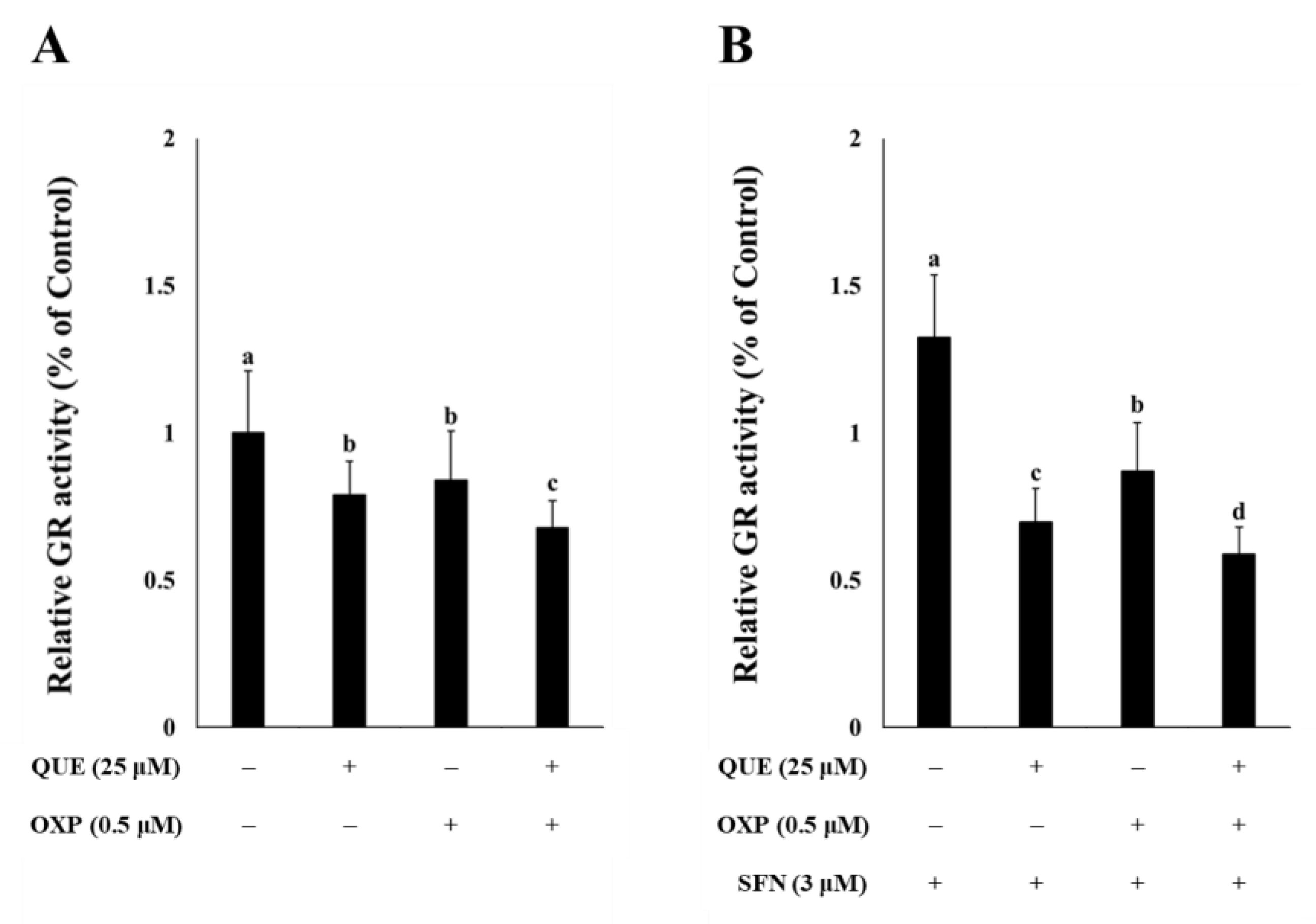
3.3. QUE Inhibited GR Activity in HCT116 Cells
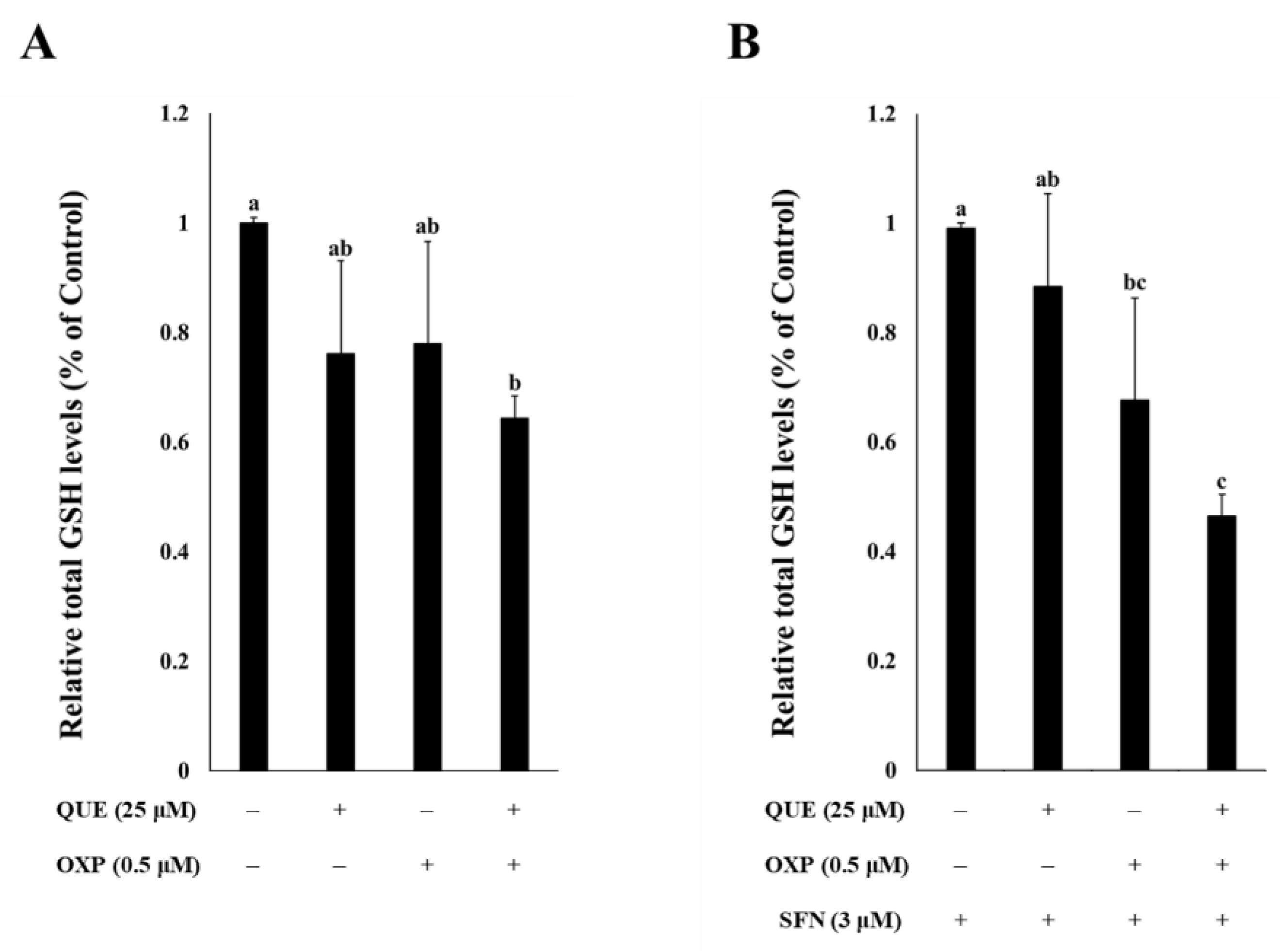
3.4. Combined Treatment of QUE and OXP Decreased Intracellular GSH Level in HCT116 Cells
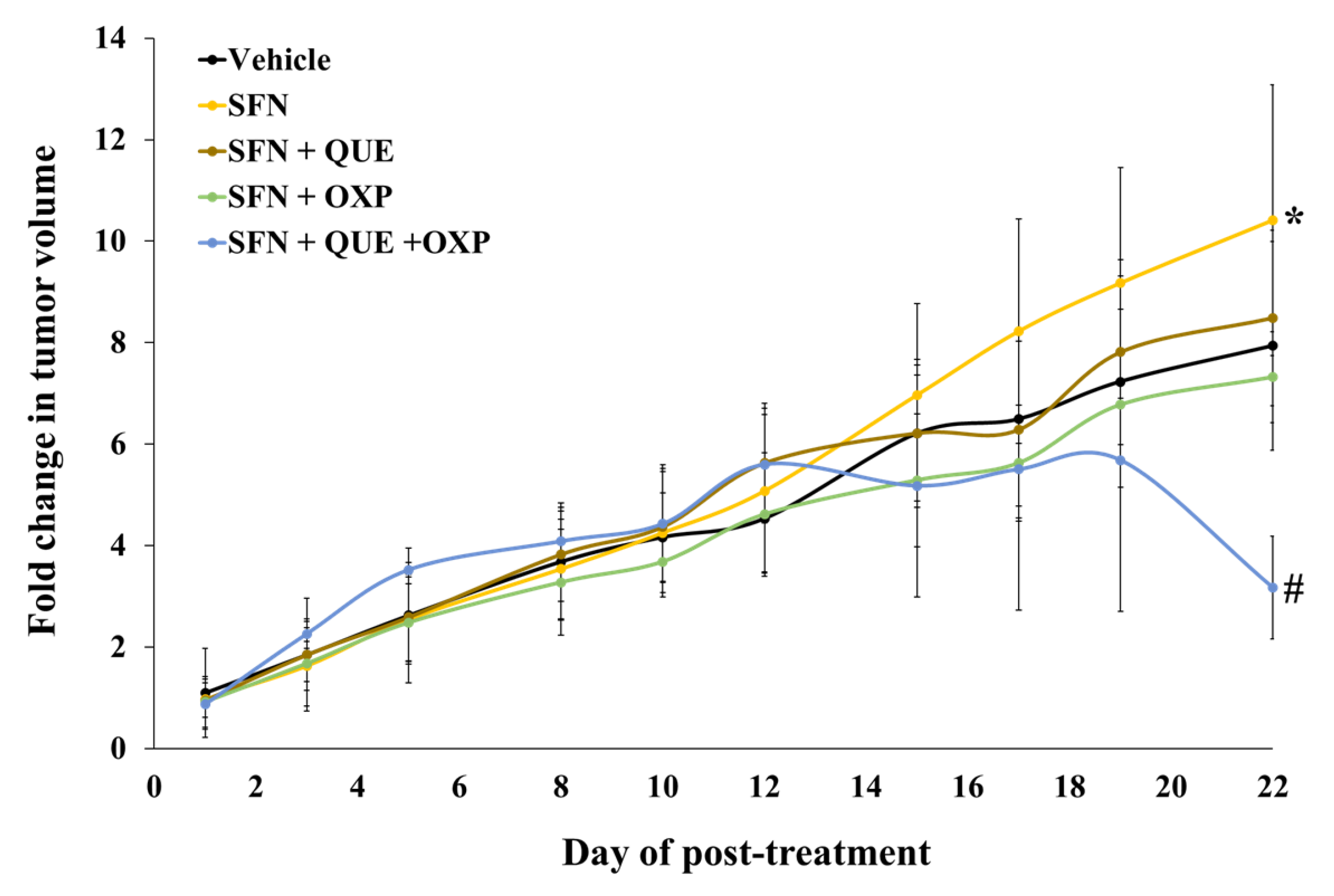
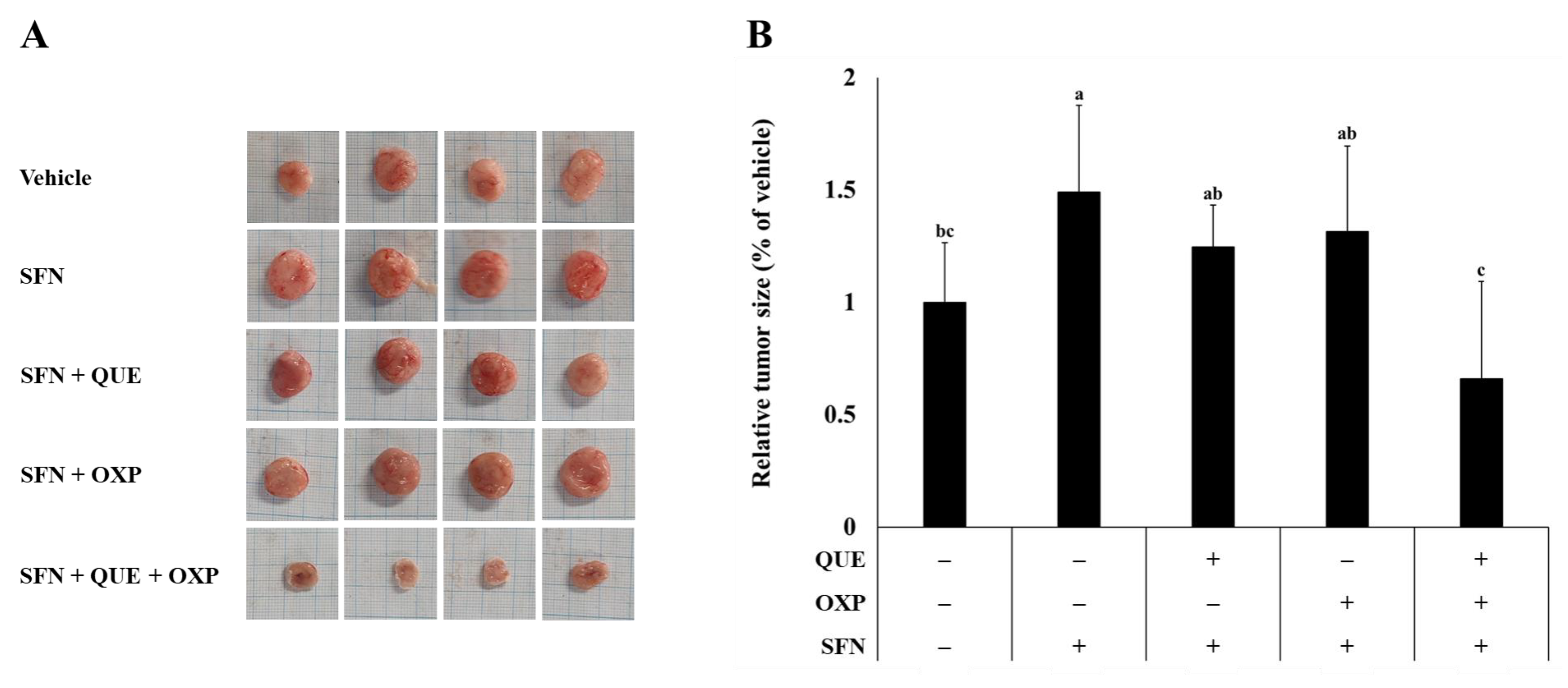
3.5. Combined Treatment of QUE and OXP Inhibited SFN-Induced HCT116 Xenograft Tumor Growth in Mice
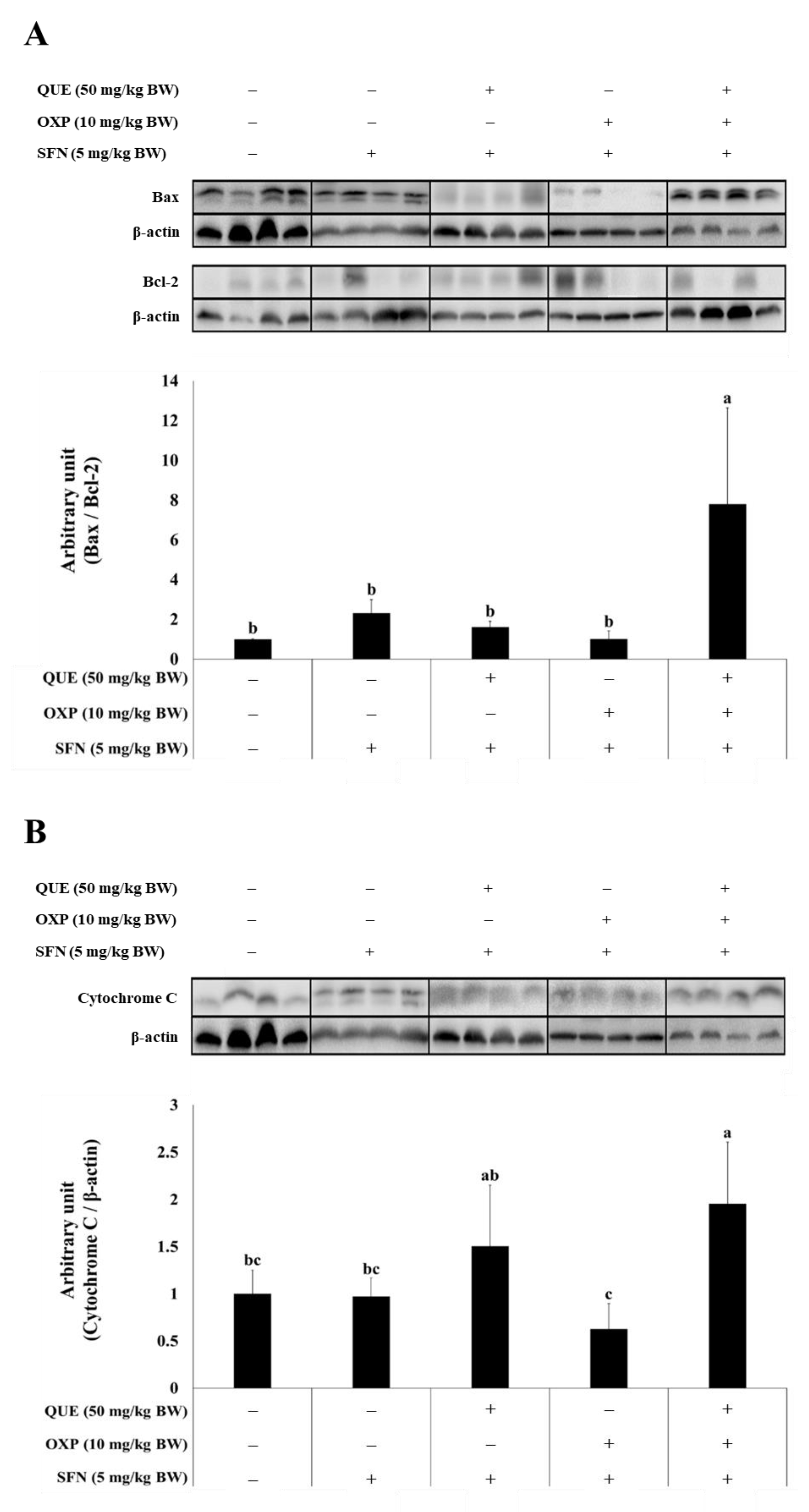
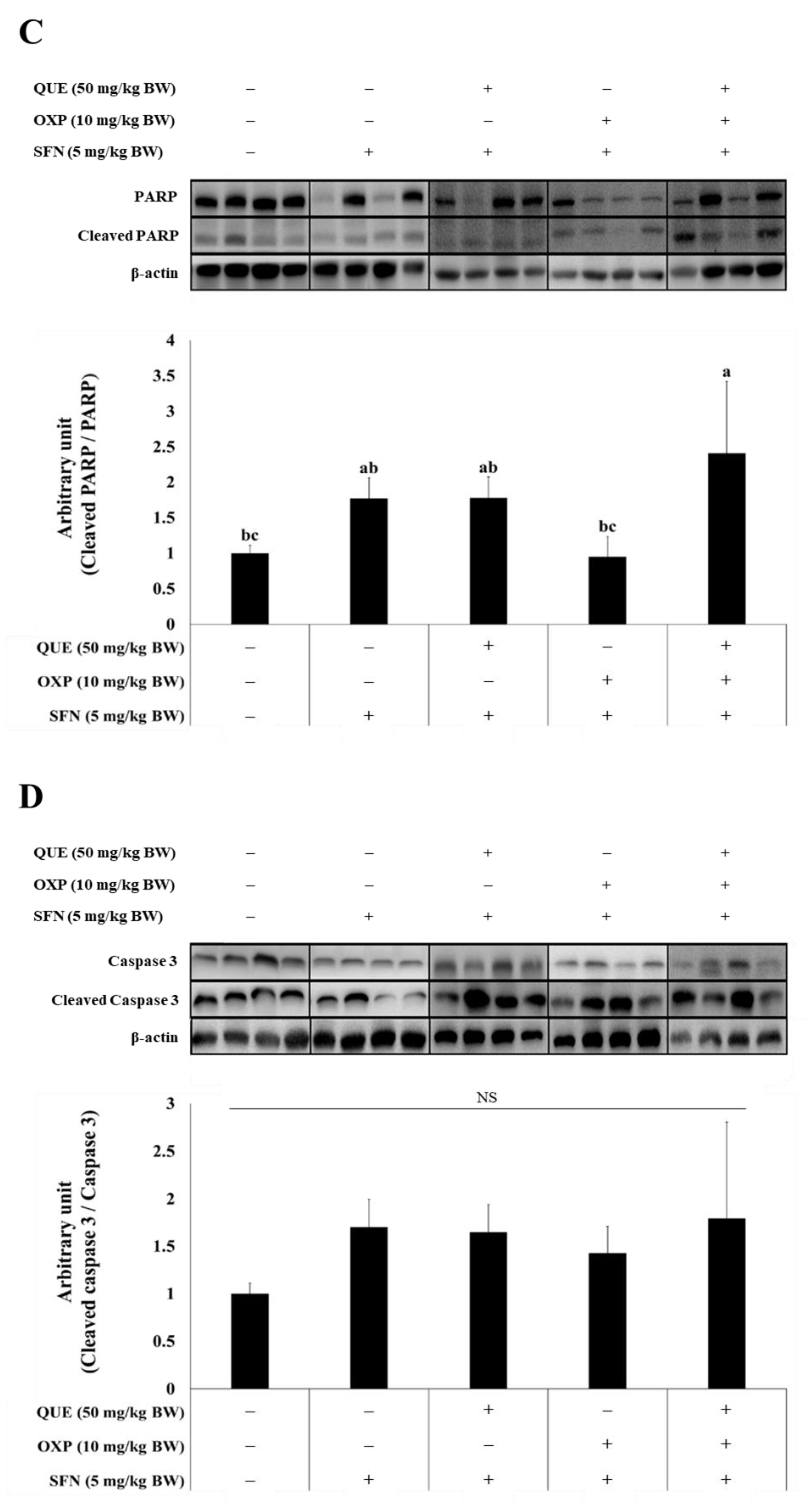
3.6. Combined Treatment of QUE and OXP Increased Pro-Apoptotic Protein Expression
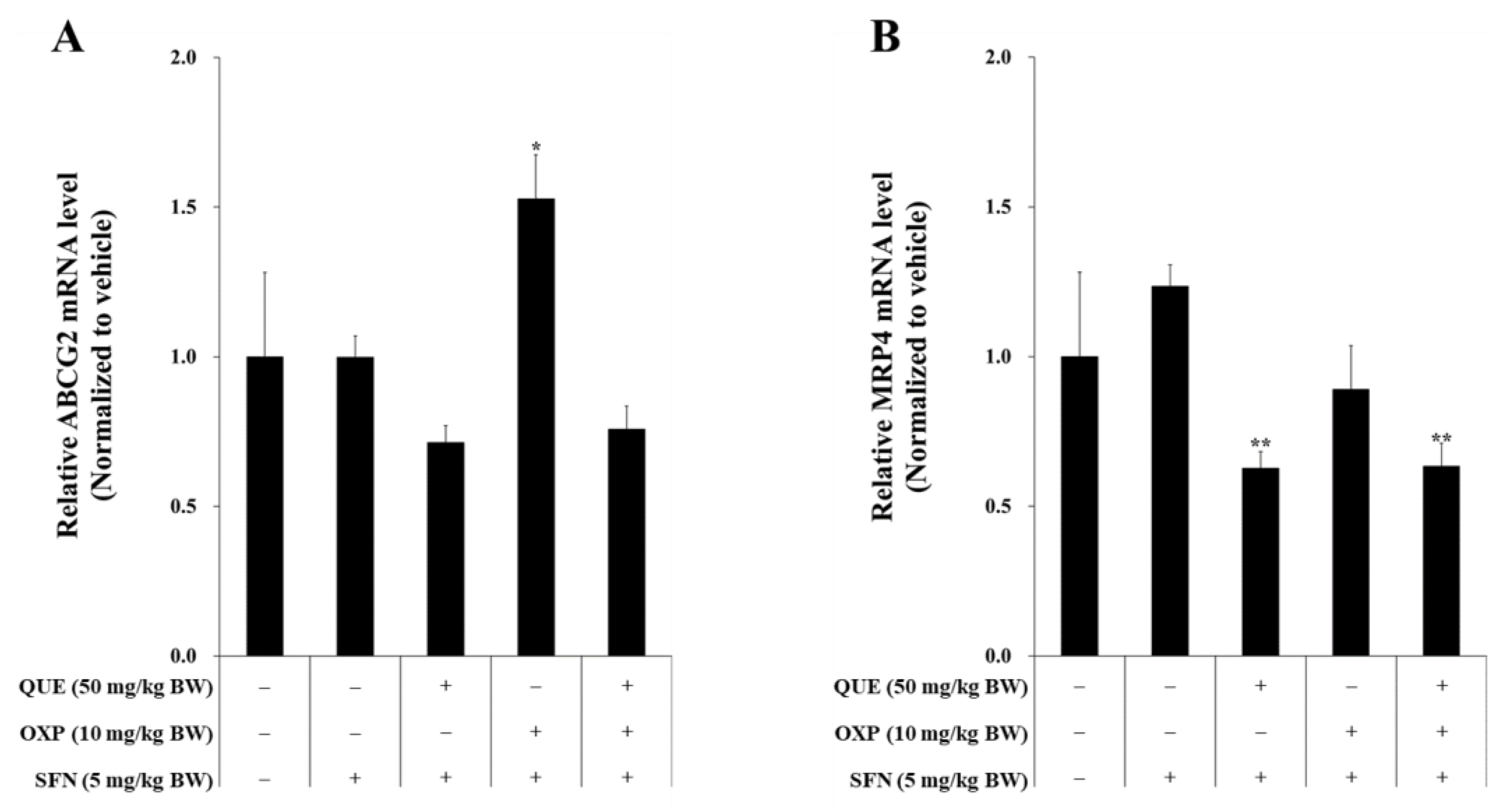
3.7. Combined Treatment of QUE and OXP Suppressed Drug-Resistance Genes in Xenograft Tumor
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.W.; Won, Y.J.; Hong, S.; Kong, H.J.; Im, J.S.; Seo, H.G. Prediction of Cancer Incidence and Mortality in Korea, 2021. Cancer Res. Treat. 2021, 53, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Misset, J.L. Oxaliplatin in practice. Br. J. Cancer 1998, 77 (Suppl. 4), 4–7. [Google Scholar] [CrossRef]
- Forgie, B.N.; Prakash, R.; Telleria, C.M. Revisiting the Anti-Cancer Toxicity of Clinically Approved Platinating Derivatives. Int. J. Mol. Sci. 2022, 23, 15410. [Google Scholar] [CrossRef]
- Arango, D.; Wilson, A.J.; Shi, Q.; Corner, G.A.; Aranes, M.J.; Nicholas, C.; Lesser, M.; Mariadason, J.M.; Augenlicht, L.H. Molecular mechanisms of action and prediction of response to oxaliplatin in colorectal cancer cells. Br. J. Cancer 2004, 91, 1931–1946. [Google Scholar] [CrossRef]
- Graham, J.; Mushin, M.; Kirkpatrick, P. Oxaliplatin. Nat. Rev. Drug Discov. 2004, 3, 11–12. [Google Scholar] [CrossRef]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhou, J.; Tong, Y. Gene signatures of drug resistance predict patient survival in colorectal cancer. Pharm. J. 2015, 15, 135–143. [Google Scholar] [CrossRef]
- Bleiberg, H. Oxaliplatin (L-OHP): A new reality in colorectal cancer. Br. J. Cancer 1998, 77 (Suppl. 4), 1–3. [Google Scholar] [CrossRef]
- Martinez-Balibrea, E.; Martinez-Cardus, A.; Gines, A.; Ruiz de Porras, V.; Moutinho, C.; Layos, L.; Manzano, J.L.; Buges, C.; Bystrup, S.; Esteller, M.; et al. Tumor-Related Molecular Mechanisms of Oxaliplatin Resistance. Mol. Cancer Ther. 2015, 14, 1767–1776. [Google Scholar] [CrossRef]
- Rafiq, R.A.; Quadri, A.; Nazir, L.A.; Peerzada, K.; Ganai, B.A.; Tasduq, S.A. A Potent Inhibitor of Phosphoinositide 3-Kinase (PI3K) and Mitogen Activated Protein (MAP) Kinase Signalling, Quercetin (3, 3′, 4′, 5, 7-Pentahydroxyflavone) Promotes Cell Death in Ultraviolet (UV)-B-Irradiated B16F10 Melanoma Cells. PLoS ONE 2015, 10, e0131253. [Google Scholar] [CrossRef]
- RiceEvans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic. Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Choi, E.J.; Lee, B.H.; Lee, K.; Chee, K.M. Long-term combined administration of quercetin and daidzein inhibits quercetin-induced suppression of glutathione antioxidant defenses. Food Chem. Toxicol. 2005, 43, 793–798. [Google Scholar] [CrossRef]
- Guller, P.; Karaman, M.; Guller, U.; Aksoy, M.; Kufrevioglu, O.I. A study on the effects of inhibition mechanism of curcumin, quercetin, and resveratrol on human glutathione reductase through in vitro and in silico approaches. J. Biomol. Struct. Dyn. 2021, 39, 1744–1753. [Google Scholar] [CrossRef]
- Gibellini, L.; Pinti, M.; Nasi, M.; De Biasi, S.; Roat, E.; Bertoncelli, L.; Cossarizza, A. Interfering with ROS Metabolism in Cancer Cells: The Potential Role of Quercetin. Cancers 2010, 2, 1288–1311. [Google Scholar] [CrossRef]
- White, C.C.; Viernes, H.; Krejsa, C.M.; Botta, D.; Kavanagh, T.J. Fluorescence-based microtiter plate assay for glutamate-cysteine ligase activity. Anal. Biochem. 2003, 318, 175–180. [Google Scholar] [CrossRef]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef]
- Zhang, J.; Hao, H.; Wu, X.; Wang, Q.; Chen, M.; Feng, Z.; Chen, H. The functions of glutathione peroxidase in ROS homeostasis and fruiting body development in Hypsizygus marmoreus. Appl. Microbiol. Biotechnol. 2020, 104, 10555–10570. [Google Scholar] [CrossRef]
- Skrzydlewska, E.; Sulkowski, S.; Koda, M.; Zalewski, B.; Kanczuga-Koda, L.; Sulkowska, M. Lipid peroxidation and antioxidant status in colorectal cancer. World J. Gastroenterol. 2005, 11, 403–406. [Google Scholar] [CrossRef]
- Jang, C.H.; Moon, N.; Lee, J.; Kwon, M.J.; Oh, J.; Kim, J.S. Luteolin Synergistically Enhances Antitumor Activity of Oxaliplatin in Colorectal Carcinoma via AMPK Inhibition. Antioxidants 2022, 11, 626. [Google Scholar] [CrossRef] [PubMed]
- Gwon, Y.; Oh, J.; Kim, J.S. Sulforaphane induces colorectal cancer cell proliferation through Nrf2 activation in a p53-dependent manner. Appl. Biol. Chem. 2020, 63, 86. [Google Scholar] [CrossRef]
- Woo, Y.; Lee, H.; Jeong, Y.S.; Shin, G.Y.; Oh, J.G.; Kim, J.S.; Oh, J. Antioxidant Potential of Selected Korean Edible Plant Extracts. BioMed Res. Int. 2017, 2017, 7695605. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.Y.; Kim, B.R.; Oh, J.; Kim, J.S. Soybean-Derived Phytoalexins Improve Cognitive Function through Activation of Nrf2/HO-1 Signaling Pathway. Int. J. Mol. Sci. 2018, 19, 268. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.; Lim, J.S.; Oh, J.; Lee, J.S.; Kim, J.S. Neuroprotective Effects of Euonymus alatus Extract on Scopolamine-Induced Memory Deficits in Mice. Antioxidants 2020, 9, 449. [Google Scholar] [CrossRef]
- Lecomte, T.; Landi, B.; Beaune, P.; Laurent-Puig, P.; Loriot, M.A. Glutathione S-transferase P1 polymorphism (Ile105Val) predicts cumulative neuropathy in patients receiving oxaliplatin-based chemotherapy. Clin. Cancer Res. 2006, 12, 3050–3056. [Google Scholar] [CrossRef]
- Theile, D.; Grebhardt, S.; Haefeli, W.E.; Weiss, J. Involvement of drug transporters in the synergistic action of FOLFOX combination chemotherapy. Biochem. Pharmacol. 2009, 78, 1366–1373. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Anand David, A.V.; Arulmoli, R.; Parasuraman, S. Overviews of Biological Importance of Quercetin: A Bioactive Flavonoid. Pharmacogn. Rev. 2016, 10, 84–89. [Google Scholar]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Vaskova, J.; Kocan, L.; Vasko, L.; Perjesi, P. Glutathione-Related Enzymes and Proteins: A Review. Molecules 2023, 28, 1447. [Google Scholar] [CrossRef]
- Elliott, A.J.; Scheiber, S.A.; Thomas, C.; Pardini, R.S. Inhibition of glutathione reductase by flavonoids. A structure-activity study. Biochem. Pharmacol. 1992, 44, 1603–1608. [Google Scholar] [CrossRef]
- Graham, M.A.; Lockwood, G.F.; Greenslade, D.; Brienza, S.; Bayssas, M.; Gamelin, E. Clinical pharmacokinetics of oxaliplatin: A critical review. Clin. Cancer Res. 2000, 6, 1205–1218. [Google Scholar]
- Burz, C.; Berindan-Neagoe, I.B.; Balacescu, O.; Tanaselia, C.; Ursu, M.; Gog, A.; Vlase, L.; Chintoanu, M.; Balacescu, L.; Leucuta, S.E.; et al. Clinical and pharmacokinetics study of oxaliplatin in colon cancer patients. J. Gastrointestin. Liver Dis. 2009, 18, 39–43. [Google Scholar]
- Levi, F.; Metzger, G.; Massari, C.; Milano, G. Oxaliplatin: Pharmacokinetics and chronopharmacological aspects. Clin. Pharmacokinet. 2000, 38, 1–21. [Google Scholar]
- Alcindor, T.; Beauger, N. Oxaliplatin: A review in the era of molecularly targeted therapy. Curr. Oncol. 2011, 18, 18–25. [Google Scholar] [CrossRef]
- Robey, R.W.; Polgar, O.; Deeken, J.; To, K.W.; Bates, S.E. ABCG2: Determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007, 26, 39–57. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef]
- Hsu, H.H.; Chen, M.C.; Baskaran, R.; Lin, Y.M.; Day, C.H.; Lin, Y.J.; Tu, C.C.; Vijaya Padma, V.; Kuo, W.W.; Huang, C.Y. Oxaliplatin resistance in colorectal cancer cells is mediated via activation of ABCG2 to alleviate ER stress induced apoptosis. J. Cell. Physiol. 2018, 233, 5458–5467. [Google Scholar] [CrossRef]
- Hlavata, I.; Mohelnikova-Duchonova, B.; Vaclavikova, R.; Liska, V.; Pitule, P.; Novak, P.; Bruha, J.; Vycital, O.; Holubec, L.; Treska, V.; et al. The role of ABC transporters in progression and clinical outcome of colorectal cancer. Mutagenesis 2012, 27, 187–196. [Google Scholar] [CrossRef]
- Giddings, E.L.; Champagne, D.P.; Wu, M.H.; Laffin, J.M.; Thornton, T.M.; Valenca-Pereira, F.; Culp-Hill, R.; Fortner, K.A.; Romero, N.; East, J.; et al. Mitochondrial ATP fuels ABC transporter-mediated drug efflux in cancer chemoresistance. Nat. Commun. 2021, 12, 2804. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Moitra, K.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Hum. Mutat. 2022, 43, 1162–1182. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Sarkadi, B.; Homolya, L.; Szakacs, G.; Varadi, A. Human multidrug resistance ABCB and ABCG transporters: Participation in a chemoimmunity defense system. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef]
- Chen, K.G.; Sikic, B.I. Molecular pathways: Regulation and therapeutic implications of multidrug resistance. Clin. Cancer Res. 2012, 18, 1863–1869. [Google Scholar] [CrossRef]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Locher, K.P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. [Google Scholar] [CrossRef]
- Sodani, K.; Patel, A.; Kathawala, R.J.; Chen, Z.S. Multidrug resistance associated proteins in multidrug resistance. Chin. J. Cancer 2012, 31, 58–72. [Google Scholar] [CrossRef]
- Pirpour Tazehkand, A.; Salehi, R.; Velaei, K.; Samadi, N. The potential impact of trigonelline loaded micelles on Nrf2 suppression to overcome oxaliplatin resistance in colon cancer cells. Mol. Biol. Rep. 2020, 47, 5817–5829. [Google Scholar] [CrossRef]
- Landriscina, M.; Maddalena, F.; Laudiero, G.; Esposito, F. Adaptation to oxidative stress, chemoresistance, and cell survival. Antioxid. Redox Signal. 2009, 11, 2701–2716. [Google Scholar] [CrossRef]
- Marioli-Sapsakou, G.K.; Kourti, M. Targeting Production of Reactive Oxygen Species as an Anticancer Strategy. Anticancer Res. 2021, 41, 5881–5902. [Google Scholar] [CrossRef]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Ohshima, K.; Morii, E. Metabolic Reprogramming of Cancer Cells during Tumor Progression and Metastasis. Metabolites 2021, 11, 28. [Google Scholar] [CrossRef]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, H.; Dong, N.; Su, X.; Duan, M.; Wei, Y.; Wei, J.; Liu, G.; Peng, Q.; Zhao, Y. Sulforaphane induces S-phase arrest and apoptosis via p53-dependent manner in gastric cancer cells. Sci. Rep. 2021, 11, 2504. [Google Scholar] [CrossRef]
- Su, X.; Jiang, X.; Meng, L.; Dong, X.; Shen, Y.; Xin, Y. Anticancer Activity of Sulforaphane: The Epigenetic Mechanisms and the Nrf2 Signaling Pathway. Oxidative Med. Cell. Longev. 2018, 2018, 5438179. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.M.; Akter, S.; Lin, C.N.; Nazzal, S. Sulforaphane as an anticancer molecule: Mechanisms of action, synergistic effects, enhancement of drug safety, and delivery systems. Arch. Pharm. Res. 2020, 43, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.; Gouge, J. Nrf2 in Cancer, Detoxifying Enzymes and Cell Death Programs. Antioxidants 2021, 10, 1030. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Johnson, J.A. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J. Biochem. Mol. Biol. 2004, 37, 139–143. [Google Scholar] [CrossRef]
- Ruhee, R.T.; Suzuki, K. The Integrative Role of Sulforaphane in Preventing Inflammation, Oxidative Stress and Fatigue: A Review of a Potential Protective Phytochemical. Antioxidants 2020, 9, 521. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Acharya, A.; Das, I.; Chandhok, D.; Saha, T. Redox regulation in cancer: A double-edged sword with therapeutic potential. Oxidative Med. Cell. Longev. 2010, 3, 23–34. [Google Scholar] [CrossRef]
- Xue, D.; Zhou, X.; Qiu, J. Emerging role of NRF2 in ROS-mediated tumor chemoresistance. Biomed. Pharmacother. 2020, 131, 110676. [Google Scholar] [CrossRef]
- Kryszczuk, M.; Kowalczuk, O. Significance of NRF2 in physiological and pathological conditions an comprehensive review. Arch. Biochem. Biophys. 2022, 730, 109417. [Google Scholar] [CrossRef]
- Xu, Y.; Han, X.; Li, Y.; Min, H.; Zhao, X.; Zhang, Y.; Qi, Y.; Shi, J.; Qi, S.; Bao, Y.; et al. Sulforaphane Mediates Glutathione Depletion via Polymeric Nanoparticles to Restore Cisplatin Chemosensitivity. ACS Nano 2019, 13, 13445–13455. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Treatment | |
|---|---|---|
| Vehicle | −SFN | Vehicle |
| SFN | +SFN (5 mg/kg BW) | Vehicle |
| QUE | QUE at 50 mg/kg BW | |
| OXP | OXP at 10 mg/kg BW | |
| QUE + OXP | QUE at 50 mg/kg BW + OXP at 10 mg/kg BW | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Jang, C.H.; Kim, Y.; Oh, J.; Kim, J.-S. Quercetin-Induced Glutathione Depletion Sensitizes Colorectal Cancer Cells to Oxaliplatin. Foods 2023, 12, 1733. https://doi.org/10.3390/foods12081733
Lee J, Jang CH, Kim Y, Oh J, Kim J-S. Quercetin-Induced Glutathione Depletion Sensitizes Colorectal Cancer Cells to Oxaliplatin. Foods. 2023; 12(8):1733. https://doi.org/10.3390/foods12081733
Chicago/Turabian StyleLee, Jinkyung, Chan Ho Jang, Yoonsu Kim, Jisun Oh, and Jong-Sang Kim. 2023. "Quercetin-Induced Glutathione Depletion Sensitizes Colorectal Cancer Cells to Oxaliplatin" Foods 12, no. 8: 1733. https://doi.org/10.3390/foods12081733