
Genetic Association Analysis of Copy Number Variations for Meat Quality in Beef Cattle
 , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Animal Populations
2.2. Phenotype Measurement
2.3. Quality Control and Detection of CNVs
2.4. CNV Compilation
2.5. CNV-Based GWAS
2.6. Region-Based GWAS
2.7. CNV Validation Using Whole-Genome Sequencing
2.8. Annotation of CNVRs and Functional Enrichment Analyses
3. Results
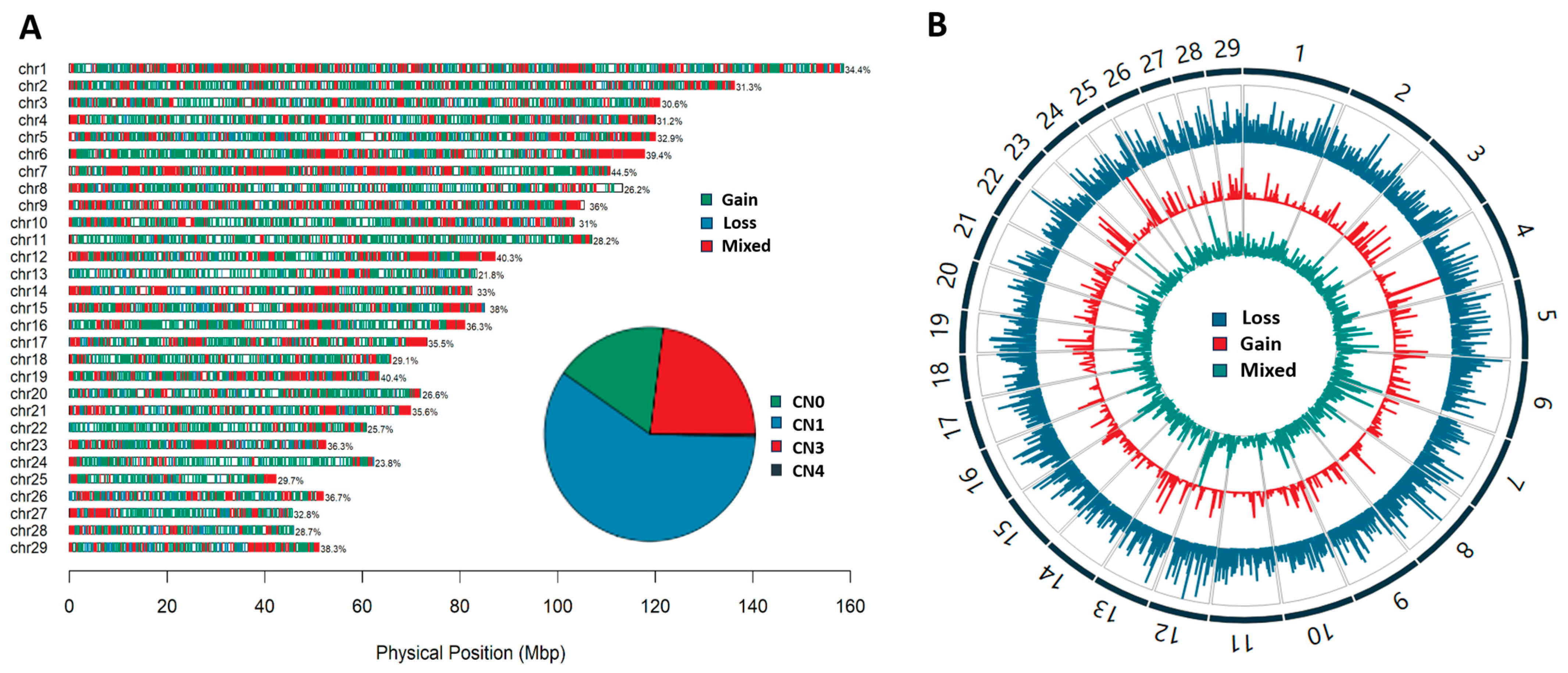
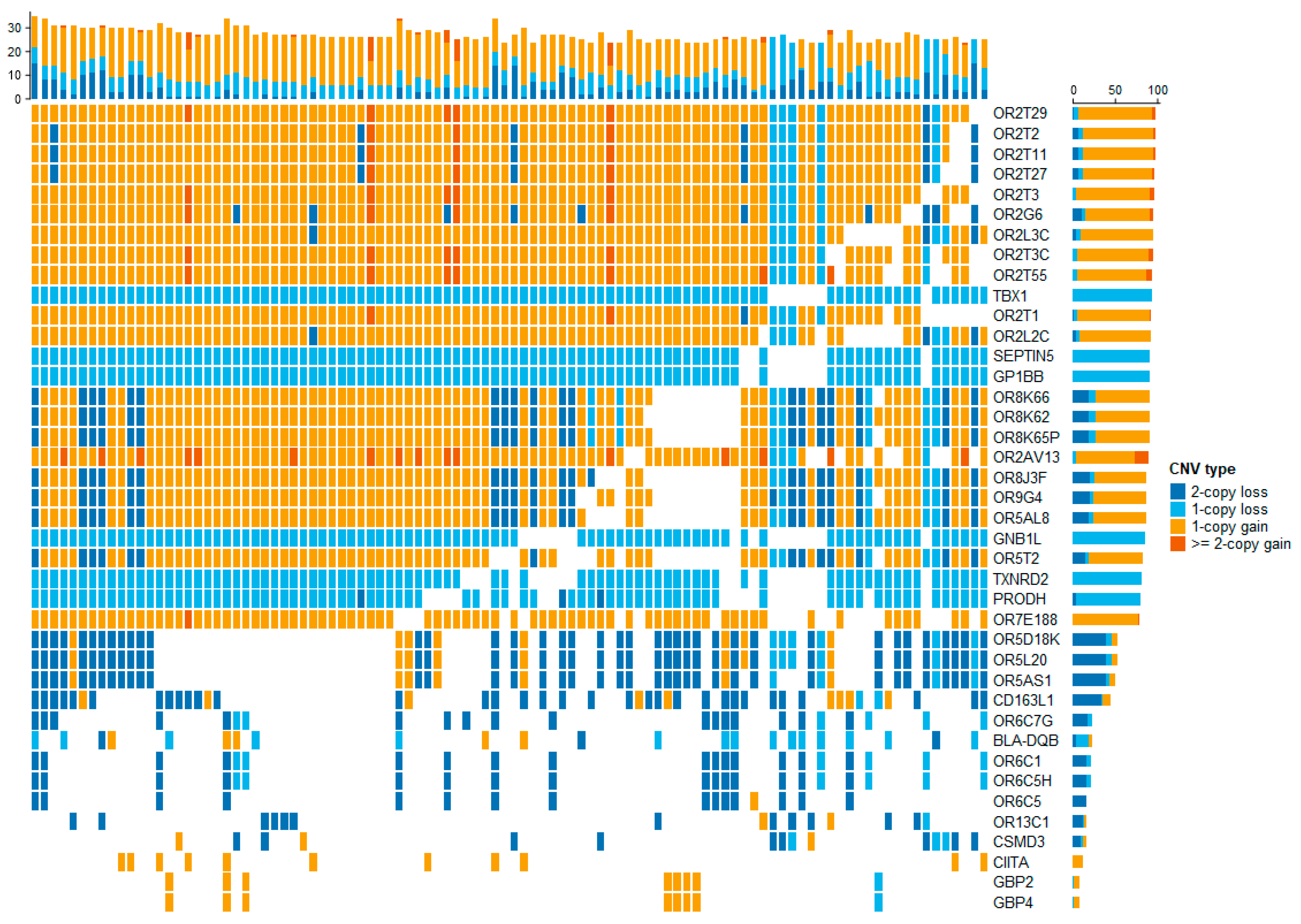
3.1. The Detection of CNVs and CNVRs
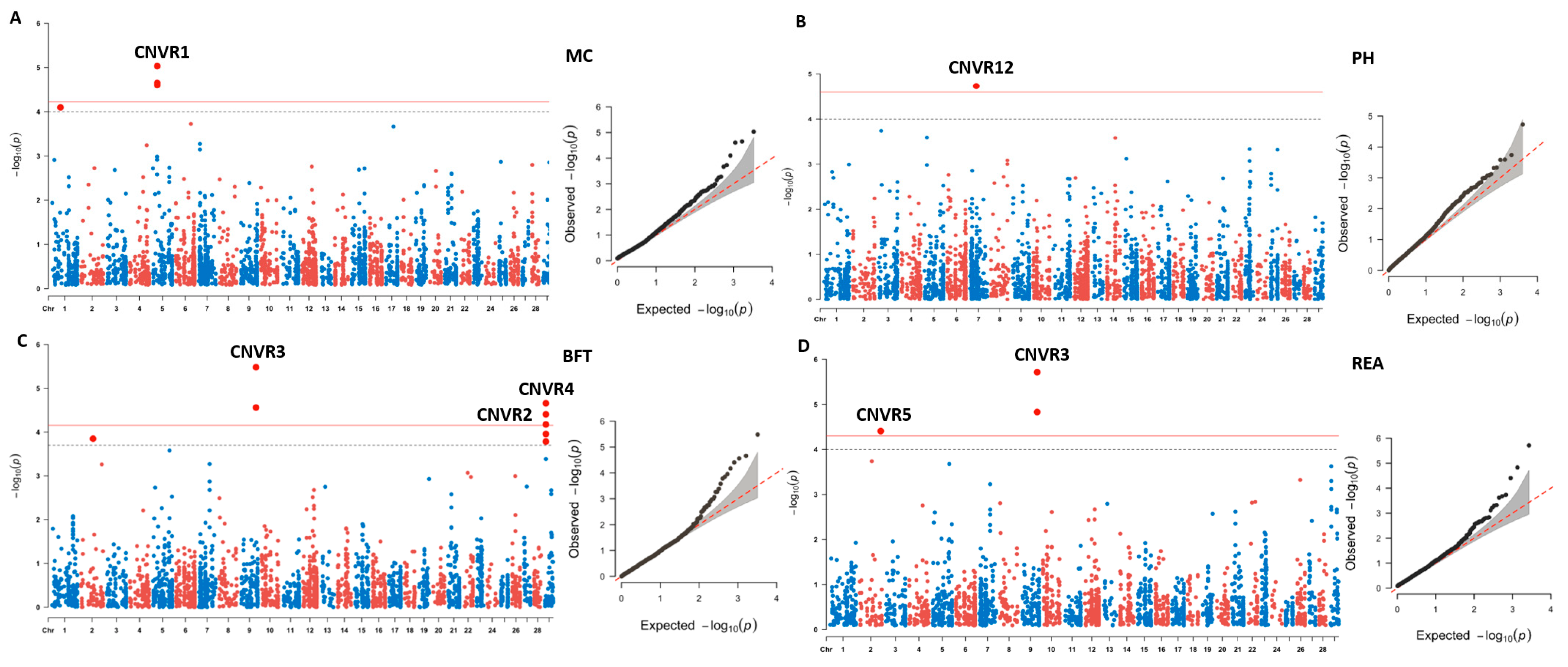
3.2. CNV-Based Association Analyses for Meat Quality
3.3. Functional Annotation
3.4. Region-Based Association Analyses of Candidate CNV Segments
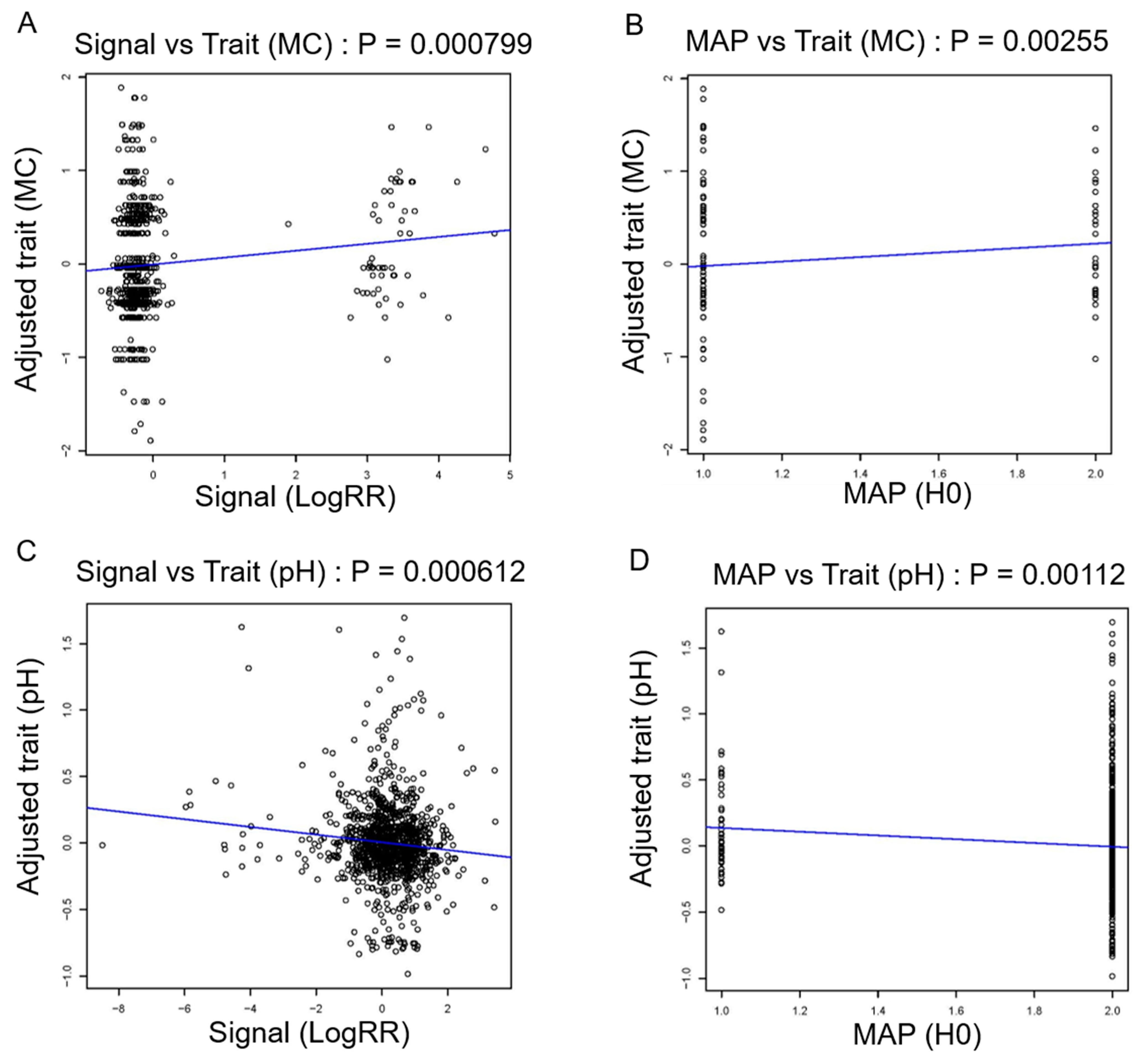
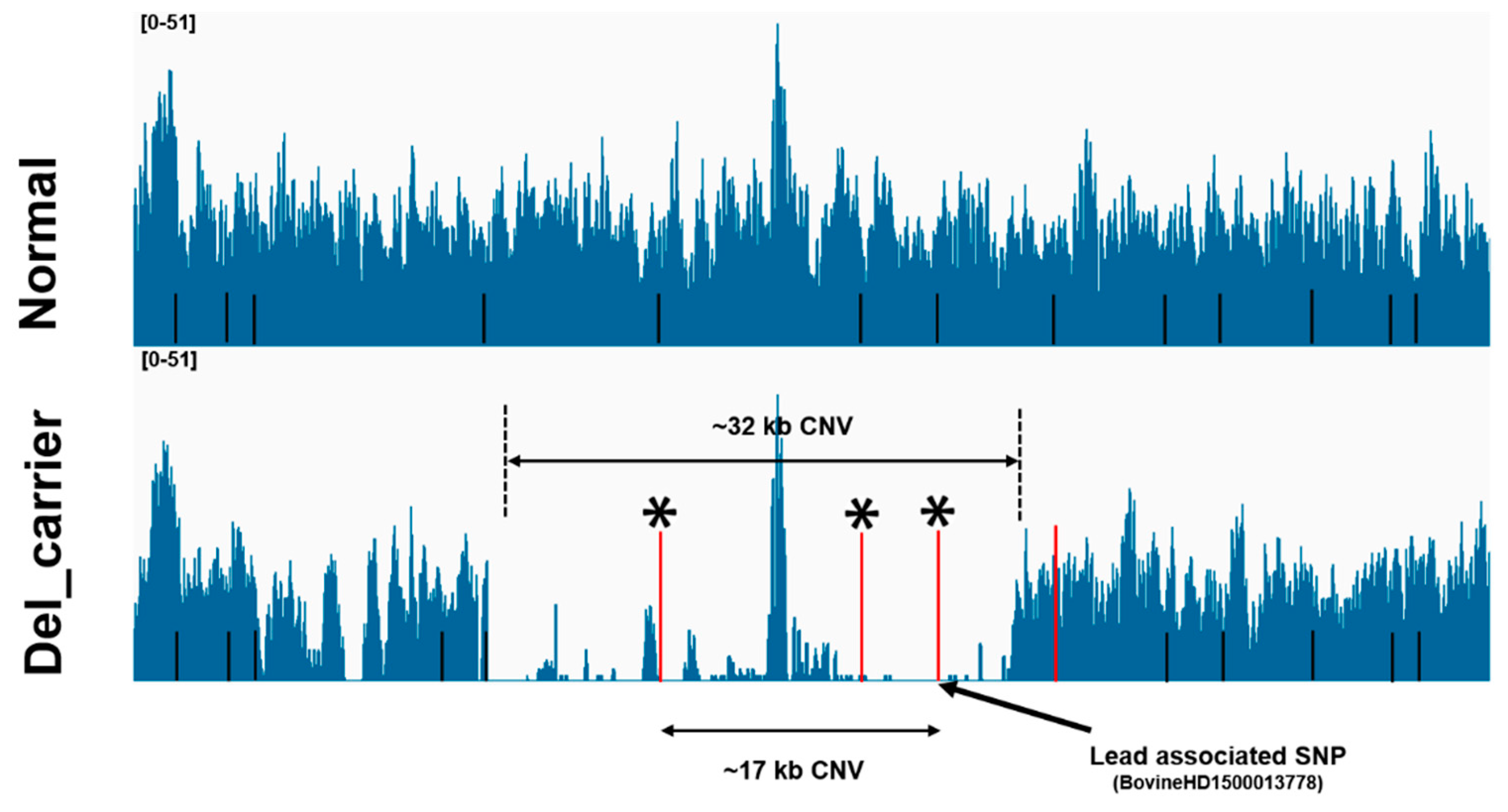
3.5. Analysis of Candidate CNVs Using Whole-Genome Sequencing Data
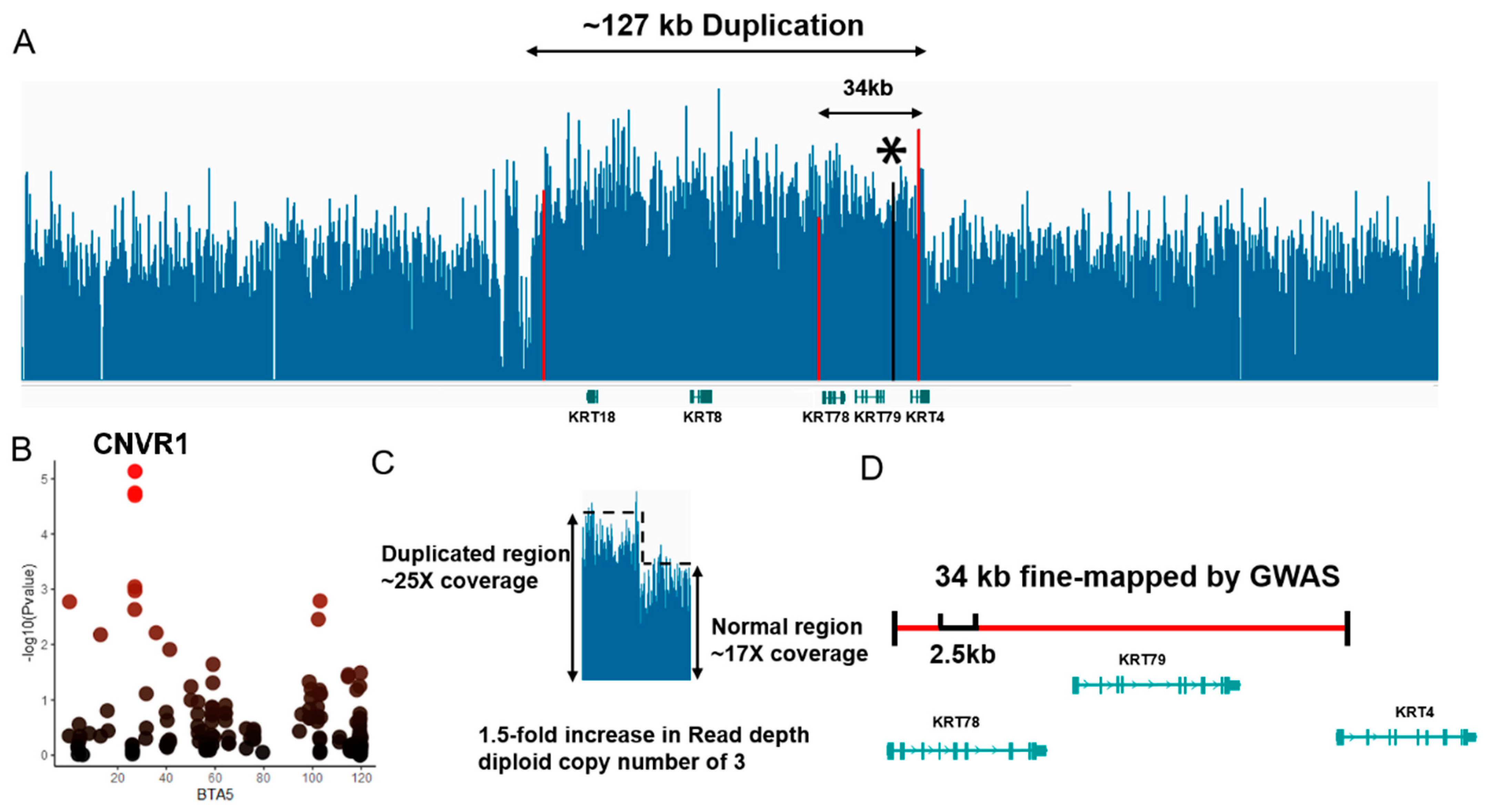
3.6. 34 kb-Duplication as a Candidate CNV Segment Related to Meat Color
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hughes, J.M.; Kearney, G.; Warner, R.D. Improving beef meat colour scores at carcass grading. J. Anim. Prod. Sci. 2014, 54, 422–429. [Google Scholar] [CrossRef]
- Żywica, R.; Modzelewska-Kapituła, M.; Banach, J.K.; Tkacz, K. Linear correlation between pH value of stimulated beef and electrical current intensity. Int. J. Food Prop. 2018, 21, 1386–1394. [Google Scholar] [CrossRef]
- Mahmood, S.; Basarab, J.A.; Dixon, W.T.; Bruce, H.L. Relationship between phenotype, carcass characteristics and the incidence of dark cutting in heifers. Meat Sci. 2016, 121, 261–271. [Google Scholar] [CrossRef]
- Mancini, R.A. 4—Meat color. In Improving the Sensory and Nutritional Quality of Fresh Meat; Kerry, J.P., Ledward, D., Eds.; Woodhead Publishing: Shaston, UK, 2009; pp. 89–110. [Google Scholar]
- Reis, M.M.; Rosenvold, K. Early on-line classification of beef carcasses based on ultimate pH by near infrared spectroscopy. Meat Sci. 2014, 96, 862–869. [Google Scholar] [CrossRef]
- Zuin, R.G.; Buzanskas, M.E.; Caetano, S.L.; Venturini, G.C.; Guidolin, D.G.; Grossi, D.A.; Chud, T.C.; Paz, C.C.; Lôbo, R.B.; Munari, D.P. Genetic analysis on growth and carcass traits in Nelore cattle. Meat Sci. 2012, 91, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.A.; Hunt, M.C. Current research in meat color. Meat Sci. 2005, 71, 100–121. [Google Scholar] [CrossRef]
- Hamoen, J.; Vollebregt, H.; Van Der Sman, R.J.M. Prediction of the time evolution of pH in meat. Food Chem. 2013, 141, 2363–2372. [Google Scholar] [CrossRef]
- Hu, Z.; Park, C.; Wu, X.-L.; Reecy, J. Animal QTLdb: An improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2012, 41, D871–D879. [Google Scholar] [CrossRef]
- Srivastava, S.; Lopez, B.I.; Heras-Saldana, S.L.; Park, J.E.; Shin, D.H.; Chai, H.H.; Park, W.; Lee, S.H.; Lim, D. Estimation of Genetic Parameters by Single-Trait and Multi-Trait Models for Carcass Traits in Hanwoo Cattle. Animals 2019, 9, 1061. [Google Scholar] [CrossRef]
- Wang, X.; Miao, J.; Chang, T.; Xia, J.; An, B.; Li, Y.; Xu, L.; Zhang, L.; Gao, X.; Li, J.; et al. Evaluation of GBLUP, BayesB and elastic net for genomic prediction in Chinese Simmental beef cattle. PLoS ONE 2019, 14, e0210442. [Google Scholar] [CrossRef]
- Marín-Garzón, N.A.; Magalhães, A.F.B.; Mota, L.F.M.; Fonseca, L.F.S.; Chardulo, L.A.L.; Albuquerque, L.G. Genome-wide association study identified genomic regions and putative candidate genes affecting meat color traits in Nellore cattle. Meat Sci. 2021, 171, 108288. [Google Scholar] [CrossRef] [PubMed]
- Santos Silva, D.B.d.; Fonseca, L.F.S.; Magalhães, A.F.B.; Muniz, M.M.M.; Baldi, F.; Ferro, J.A.; Chardulo, L.A.L.; Pinheiro, D.G.; Albuquerque, L.G.d. Transcriptome profiling of muscle in Nelore cattle phenotypically divergent for the ribeye muscle area. Genomics 2020, 112, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yang, R.; Lu, X.; Liu, Y.; He, W.; Li, Y.; Yu, H.; Qin, L.; Cao, Y.; Zhao, Z.; et al. RNA-Seq Analysis Identifies Differentially Expressed Genes in the Longissimus dorsi of Wagyu and Chinese Red Steppe Cattle. Int. J. Mol. Sci. 2022, 24, 387. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, J.; Wang, X.; Han, L.; Yang, Y.; Wang, Q.; Yu, Q. Metagenomic and Transcriptomic Analyses Reveal the Differences and Associations between the Gut Microbiome and Muscular Genes in Angus and Chinese Simmental Cattle. Front. Microbiol. 2022, 13, 815915. [Google Scholar] [CrossRef]
- Wang, Z.; An, X.; Yang, Y.; Zhang, L.; Jiao, T.; Zhao, S. Comprehensive Analysis of the Longissimus Dorsi Transcriptome and Metabolome Reveals the Regulatory Mechanism of Different Varieties of Meat Quality. J. Agric. Food Chem. 2023, 71, 1234–1245. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, Y.; Niu, Q.; Zheng, X.; Zhang, T.; Wang, Z.; Xu, L.; Zhu, B.; Gao, X.; Zhang, L.; et al. Runs of homozygosity analysis reveals consensus homozygous regions affecting production traits in Chinese Simmental beef cattle. BMC Genom. 2021, 22, 678. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Li, K.; Chang, T.; An, B.; Liang, M.; Deng, T.; Cao, S.; Du, Y.; Cai, W.; Gao, X.; et al. Integrating genomics and transcriptomics to identify candidate genes for subcutaneous fat deposition in beef cattle. Genomics 2022, 114, 110406. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Li, T.; Su, M.; Wang, H.; Li, Q.; Lang, X.; Ma, Y. Identification of copy number variation in Tibetan sheep using whole genome resequencing reveals evidence of genomic selection. BMC Genom. 2023, 24, 555. [Google Scholar] [CrossRef]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef]
- Watanabe, K.; Stringer, S.; Frei, O.; Umićević Mirkov, M.; de Leeuw, C.; Polderman, T.J.C.; van der Sluis, S.; Andreassen, O.A.; Neale, B.M.; Posthuma, D. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019, 51, 1339–1348. [Google Scholar] [CrossRef]
- Canela-Xandri, O.; Rawlik, K.; Tenesa, A. An atlas of genetic associations in UK Biobank. Nat. Genet. 2018, 50, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Wu, Y.; Zhou, X.; Xia, J.; Zhang, W.; Song, Y.; Liu, F.; Chen, Y.; Zhang, L.; Gao, X.; et al. Pathway-Based Genome-Wide Association Studies for Two Meat Production Traits in Simmental Cattle. Sci. Rep. 2015, 5, 18389. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Fan, H.; Chang, T.; Xu, L.; Zhang, W.; Song, Y.; Zhu, B.; Zhang, L.; Gao, X.; Chen, Y.; et al. Searching for new loci and candidate genes for economically important traits through gene-based association analysis of Simmental cattle. Sci. Rep. 2017, 7, 42048. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Qi, X.; Wu, Y.; Zhu, B.; Xu, L.; Zhang, L.; Gao, X.; Chen, Y.; Li, J.; Gao, H. Genome-wide association study identifies loci and candidate genes for meat quality traits in Simmental beef cattle. Mamm. Genome 2016, 27, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef]
- Baker, M. Structural variation: The genome’s hidden architecture. Nat. Methods 2012, 9, 133–137. [Google Scholar] [CrossRef]
- Hollis, B.; Day, F.R.; Busch, A.S.; Thompson, D.J.; Soares, A.L.G.; Timmers, P.; Kwong, A.; Easton, D.F.; Joshi, P.K.; Timpson, N.J.; et al. Genomic analysis of male puberty timing highlights shared genetic basis with hair colour and lifespan. Nat. Commun. 2020, 11, 1536. [Google Scholar] [CrossRef]
- Aerts, E.; Beckers, S.; Zegers, D.; Van Hoorenbeeck, K.; Massa, G.; Verrijken, A.; Verhulst, S.L.; Van Gaal, L.F.; Van Hul, W. CNV analysis and mutation screening indicate an important role for the NPY4R gene in human obesity. Obesity 2016, 24, 970–976. [Google Scholar] [CrossRef]
- Wolujewicz, P.; Aguiar-Pulido, V.; AbdelAleem, A.; Nair, V.; Thareja, G.; Suhre, K.; Shaw, G.M.; Finnell, R.H.; Elemento, O.; Ross, M.E. Genome-wide investigation identifies a rare copy-number variant burden associated with human spina bifida. Genet. Med. 2021, 23, 1211–1218. [Google Scholar] [CrossRef]
- Verbitsky, M.; Krithivasan, P.; Batourina, E.; Khan, A.; Graham, S.E.; Marasà, M.; Kim, H.; Lim, T.Y.; Weng, P.L.; Sánchez-Rodríguez, E.; et al. Copy Number Variant Analysis and Genome-wide Association Study Identify Loci with Large Effect for Vesicoureteral Reflux. J. Am. Soc. Nephrol. JASN 2021, 32, 805–820. [Google Scholar] [CrossRef]
- Montanucci, L.; Lewis-Smith, D.; Collins, R.L.; Niestroj, L.-M.; Parthasarathy, S.; Xian, J.; Ganesan, S.; Macnee, M.; Brünger, T.; Thomas, R.H.; et al. Genome-wide identification and phenotypic characterization of seizure-associated copy number variations in 741,075 individuals. Nat. Commun. 2023, 14, 4392. [Google Scholar] [CrossRef]
- Qiu, Y.; Ding, R.; Zhuang, Z.; Wu, J.; Yang, M.; Zhou, S.; Ye, Y.; Geng, Q.; Xu, Z.; Huang, S.; et al. Genome-wide detection of CNV regions and their potential association with growth and fatness traits in Duroc pigs. BMC Genom. 2021, 22, 332. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Zhuang, Z.; Qiu, Y.; Wang, X.; Wu, J.; Zhou, S.; Ruan, D.; Xu, C.; Hong, L.; Gu, T.; et al. A composite strategy of genome-wide association study and copy number variation analysis for carcass traits in a Duroc pig population. BMC Genom. 2022, 23, 590. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, S.; Gholizadeh, M.; Rahimi-Mianji, G.; Moradi, M.H.; Costilla, R.; Moore, S.; Di Gerlando, R. Genome-wide identification of copy number variation and association with fat deposition in thin and fat-tailed sheep breeds. Sci. Rep. 2022, 12, 8834. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.; Saif, R.; Jagannathan, V.; Schmocker, C.; Zeindler, F.; Bangerter, E.; Herren, U.; Posantzis, D.; Bulut, Z.; Ammann, P.; et al. Selection signatures in goats reveal copy number variants underlying breed-defining coat color phenotypes. PLoS Genet. 2019, 15, e1008536. [Google Scholar] [CrossRef]
- Weich, K.; Affolter, V.; York, D.; Rebhun, R.; Grahn, R.; Kallenberg, A.; Bannasch, D. Pigment Intensity in Dogs is Associated with a Copy Number Variant Upstream of KITLG. Genes 2020, 11, 75. [Google Scholar] [CrossRef]
- Zhou, Y.; Utsunomiya, Y.T.; Xu, L.; Hayel, H.A.; Bickhart, D.M.; Alexandre, P.A.; Rosen, B.D.; Schroeder, S.G.; Carvalheiro, R.; de Rezende Neves, H.H.; et al. Genome-wide CNV analysis reveals variants associated with growth traits in Bos indicus. BMC Genom. 2016, 17, 419. [Google Scholar] [CrossRef]
- Liu, M.; Fang, L.; Liu, S.; Pan, M.G.; Seroussi, E.; Cole, J.B.; Ma, L.; Chen, H.; Liu, G.E. Array CGH-based detection of CNV regions and their potential association with reproduction and other economic traits in Holsteins. BMC Genom. 2019, 20, 181. [Google Scholar] [CrossRef]
- Xu, L.; Yang, L.; Wang, L.; Zhu, B.; Chen, Y.; Gao, H.; Gao, X.; Zhang, L.; Liu, G.E.; Li, J. Probe-based association analysis identifies several deletions associated with average daily gain in beef cattle. BMC Genom. 2019, 20, 31. [Google Scholar] [CrossRef]
- Yang, L.; Niu, Q.; Zhang, T.; Zhao, G.; Zhu, B.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Liu, G.E.; et al. Genomic sequencing analysis reveals copy number variations and their associations with economically important traits in beef cattle. Genomics 2021, 113, 812–820. [Google Scholar] [CrossRef]
- Liu, M.; Woodward-Greene, J.; Kang, X.; Pan, M.G.; Rosen, B.; Van Tassell, C.P.; Chen, H.; Liu, G.E. Genome-wide CNV analysis revealed variants associated with growth traits in African indigenous goats. Genomics 2020, 112, 1477–1480. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, F.; Mukiibi, R.; Chen, L.; Vinsky, M.; Plastow, G.; Basarab, J.; Stothard, P.; Li, C. Genetic architecture of quantitative traits in beef cattle revealed by genome wide association studies of imputed whole genome sequence variants: II: Carcass merit traits. BMC Genom. 2020, 21, 38. [Google Scholar] [CrossRef] [PubMed]
- Bedhane, M.; van der Werf, J.; Gondro, C.; Duijvesteijn, N.; Lim, D.; Park, B.; Park, M.N.; Hee, R.S.; Clark, S. Genome-Wide Association Study of Meat Quality Traits in Hanwoo Beef Cattle Using Imputed Whole-Genome Sequence Data. Front. Genet. 2019, 10, 1235. [Google Scholar] [CrossRef]
- Grigoletto, L.; Ferraz, J.B.S.; Oliveira, H.R.; Eler, J.P.; Bussiman, F.O.; Abreu Silva, B.C.; Baldi, F.; Brito, L.F. Genetic Architecture of Carcass and Meat Quality Traits in Montana Tropical (R) Composite Beef Cattle. Front. Genet. 2020, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Mateescu, R.G.; Garrick, D.J.; Reecy, J.M. Network Analysis Reveals Putative Genes Affecting Meat Quality in Angus Cattle. Front. Genet. 2017, 8, 171. [Google Scholar] [CrossRef]
- Silva, V.H.; Regitano, L.C.; Geistlinger, L.; Pértille, F.; Giachetto, P.F.; Brassaloti, R.A.; Morosini, N.S.; Zimmer, R.; Coutinho, L.L. Genome-Wide Detection of CNVs and Their Association with Meat Tenderness in Nelore Cattle. PLoS ONE 2016, 11, e0157711. [Google Scholar] [CrossRef]
- Xu, L.; Gao, N.; Wang, Z.; Xu, L.; Liu, Y.; Chen, Y.; Xu, L.; Gao, X.; Zhang, L.; Gao, H.; et al. Incorporating Genome Annotation Into Genomic Prediction for Carcass Traits in Chinese Simmental Beef Cattle. Front. Genet. 2020, 11, 481. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, L.; Lopdell, T.J.; Garrick, D.J.; Shi, Y. HandyCNV: Standardized Summary, Annotation, Comparison, and Visualization of Copy Number Variant, Copy Number Variation Region, and Runs of Homozygosity. Front. Genet. 2021, 12, 731355. [Google Scholar] [CrossRef]
- da Silva, V.; Ramos, M.; Groenen, M.; Crooijmans, R.; Johansson, A.; Regitano, L.; Coutinho, L.; Zimmer, R.; Waldron, L.; Geistlinger, L. CNVRanger: Association analysis of CNVs with gene expression and quantitative phenotypes. Bioinformatics 2020, 36, 972–973. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.; Plagnol, V.; Fitzgerald, T.; Redon, R.; Marchini, J.; Clayton, D.; Hurles, M.E. A robust statistical method for case-control association testing with copy number variation. Nat. Genet. 2008, 40, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Huang, Y.; Li, Y.; Wang, X.; Yu, J.; Cai, Y.; Zheng, Z.; Li, R.; Zhang, S.; Chen, N.; Asadollahpour Nanaei, H.; et al. An atlas of CNV maps in cattle, goat and sheep. Science China. Life Sci. 2021, 64, 1747–1764. [Google Scholar] [CrossRef]
- Fonseca, P.A.S.; Suárez-Vega, A.; Marras, G.; Cánovas, Á. GALLO: An R package for genomic annotation and integration of multiple data sources in livestock for positional candidate loci. GigaScience 2020, 9, giaa149. [Google Scholar] [CrossRef]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 2021, 50, D956–D961. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef]
- Xu, L.; Cole, J.B.; Bickhart, D.M.; Hou, Y.; Song, J.; VanRaden, P.M.; Sonstegard, T.S.; Van Tassell, C.P.; Liu, G.E. Genome wide CNV analysis reveals additional variants associated with milk production traits in Holsteins. BMC Genom. 2014, 15, 683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Guo, X.; Zhang, Y.; Wen, Y.; Wang, W.; Wang, S.; Yang, T.; Shen, H.; Chen, X.; Tian, Q.; et al. Genome-wide copy number variation study and gene expression analysis identify ABI3BP as a susceptibility gene for Kashin-Beck disease. Hum. Genet. 2014, 133, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.C.; da Silva, V.H.; Goes, C.P.; Moreira, G.C.M.; Godoy, T.F.; Ibelli, A.M.G.; Peixoto, J.O.; Cantão, M.E.; Ledur, M.C.; de Rezende, F.M.; et al. Genome-wide detection of CNVs and their association with performance traits in broilers. BMC Genom. 2021, 22, 354. [Google Scholar] [CrossRef] [PubMed]
- González, J.R.; Subirana, I.; Escaramís, G.; Peraza, S.; Cáceres, A.; Estivill, X.; Armengol, L. Accounting for uncertainty when assessing association between copy number and disease: A latent class model. BMC Bioinform. 2009, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Ionita-Laza, I.; Perry, G.H.; Raby, B.A.; Klanderman, B.; Lee, C.; Laird, N.M.; Weiss, S.T.; Lange, C. On the analysis of copy-number variations in genome-wide association studies: A translation of the family-based association test. Genet. Epidemiol. 2008, 32, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Stranger, B.E.; Forrest, M.S.; Dunning, M.; Ingle, C.E.; Beazley, C.; Thorne, N.; Redon, R.; Bird, C.P.; de Grassi, A.; Lee, C.; et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 2007, 315, 848–853. [Google Scholar] [CrossRef]
- McCarroll, S.A.; Altshuler, D.M. Copy-number variation and association studies of human disease. Nat. Genet. 2007, 39, S37–S42. [Google Scholar] [CrossRef]
- Chao, S.C.; Tsai, Y.M.; Yang, M.H.; Lee, J.Y. A novel mutation in the keratin 4 gene causing white sponge naevus. Br. J. Dermatol. 2003, 148, 1125–1128. [Google Scholar] [CrossRef]
- Veniaminova, N.A.; Grachtchouk, M.; Doane, O.J.; Peterson, J.K.; Quigley, D.A.; Lull, M.V.; Pyrozhenko, D.V.; Nair, R.R.; Patrick, M.T.; Balmain, A.; et al. Niche-Specific Factors Dynamically Regulate Sebaceous Gland Stem Cells in the Skin. Dev. Cell 2019, 51, 326–340.e324. [Google Scholar] [CrossRef]
- Dai, B.; Liang, H.; Guo, D.D.; Bi, Z.W.; Yuan, J.L.; Jin, Y.; Huan, L.; Guo, X.D.; Cang, M.; Liu, D.J. The Overexpression of Tβ4 in the Hair Follicle Tissue of Alpas Cashmere Goats Increases Cashmere Yield and Promotes Hair Follicle Development. Animals 2019, 10, 75. [Google Scholar] [CrossRef]
- Bao, Q.; Zhang, X.; Bao, P.; Liang, C.; Guo, X.; Yin, M.; Chu, M.; Yan, P. Genome-wide identification, characterization, and expression analysis of keratin genes (KRTs) family in yak (Bos grunniens). Gene 2022, 818, 146247. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, C.; Peng, W.; Du, H.; Xi, Y.; Xu, Z. RBM24 is required for mouse hair cell development through regulating pre-mRNA alternative splicing and mRNA stability. J. Cell. Physiol. 2023, 238, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Krey, J.F.; Chatterjee, P.; Halford, J.; Cunningham, C.L.; Perrin, B.J.; Barr-Gillespie, P.G. Control of stereocilia length during development of hair bundles. PLoS Biol. 2023, 21, e3001964. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Qu, K.; Ma, Z.; Zhan, J.; Zhang, F.; Shen, J.; Ning, Q.; Jia, P.; Zhang, J.; Chen, N.; et al. Genome-Wide Association Study Identifies Genomic Loci Associated With Neurotransmitter Concentration in Cattle. Front. Genet. 2020, 11, 139. [Google Scholar] [CrossRef]
- Zlobin, A.S.; Volkova, N.A.; Zinovieva, N.A.; Iolchiev, B.S.; Bagirov, V.A.; Borodin, P.M.; Axenovich, T.I.; Tsepilov, Y.A. Loci Associated with Negative Heterosis for Viability and Meat Productivity in Interspecific Sheep Hybrids. Animals 2023, 13, 184. [Google Scholar] [CrossRef]
- Hu, L.; Zhang, L.; Li, Q.; Liu, H.; Xu, T.; Zhao, N.; Han, X.; Xu, S.; Zhao, X.; Zhang, C. Genome-wide analysis of CNVs in three populations of Tibetan sheep using whole-genome resequencing. Front. Genet. 2022, 13, 971464. [Google Scholar] [CrossRef]
- Floris, C.; Rassu, S.; Boccone, L.; Gasperini, D.; Cao, A.; Crisponi, L. Two patients with balanced translocations and autistic disorder: CSMD3 as a candidate gene for autism found in their common 8q23 breakpoint area. Eur. J. Hum. Genet. 2008, 16, 696–704. [Google Scholar] [CrossRef]
- Sarowar, T.; Chhabra, R.; Vilella, A.; Boeckers, T.M.; Zoli, M.; Grabrucker, A.M. Activity and circadian rhythm influence synaptic Shank3 protein levels in mice. J. Neurochem. 2016, 138, 887–895. [Google Scholar] [CrossRef]
- Zhu, C.; Chen, W.; Cui, H.; Huang, Z.; Ding, R.; Li, N.; Wang, Q.; Wu, F.; Zhao, Y.; Cong, X. TRIM64 promotes ox-LDL-induced foam cell formation, pyroptosis, and inflammation in THP-1-derived macrophages by activating a feedback loop with NF-κB via IκBα ubiquitination. Cell Biol. Toxicol. 2022, 39, 607–620. [Google Scholar] [CrossRef]
- Peng, H.; Liu, G.; Bao, Y.; Zhang, X.; Zhou, L.; Huang, C.; Song, Z.; Cao, S.; Dang, S.; Zhang, J.; et al. Prognostic Factors of Colorectal Cancer: A Comparative Study on Patients With or wthout Liver wetastasis. Front. Oncol. 2021, 11, 626190. [Google Scholar] [CrossRef]
- Giuffra, E.; Tuggle, C.K. Functional Annotation of Animal Genomes (FAANG): Current Achievements and Roadmap. Annu. Rev. Anim. Biosci. 2019, 7, 65–88. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.; Archibald, A.L.; Bottema, C.D.; Brauning, R.; Burgess, S.C.; Burt, D.W.; Casas, E.; Cheng, H.H.; Clarke, L.; Couldrey, C.; et al. Coordinated international action to accelerate genome-to-phenome with FAANG, the Functional Annotation of Animal Genomes project. Genome Biol. 2015, 16, 57. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | CNV Segment | BTA | Location | Type | p Value | FDR | Candidate Gene |
|---|---|---|---|---|---|---|---|
| MC | CNVR1 | 5 | 27,085,360–27,119,759 | Duplication | 9.28 × 10−06 | 0.033 | KRT4, KRT78, KRT79 |
| BFT | CNVR2 | 29 | 5,171,306–5,281,082 | Duplication | 2.20 × 10−05 | 0.030 | |
| CNVR3 | 9 | 92,831,482–92,843,287 | Duplication | 3.30 × 10−06 | 0.006 | ||
| CNVR4 | 29 | 5,287,673–5,295,854 | Duplication | 9.28 × 10−06 | 0.032 | TRIM77, TRIM64 | |
| REA | CNVR3 | 9 | 92,831,482–92,843,287 | Duplication | 1.93 × 10−06 | 0.006 | |
| CNVR5 | 2 | 129,599,911–129,603,637 | Deletion | 6.22 × 10−05 | 0.042 | ||
| MS | CNVR6 | 9 | 30,539,303–30,579,950 | Deletion | 6.22 × 10−05 | 0.087 | |
| CNVR7 | 26 | 4,750,372–4,775,984 | Deletion | 1.23 × 10−04 | 0.099 | PCDH15 | |
| CNVR8 | 20 | 60,391,422–60,425,423 | Both | 1.15 × 10−04 | 0.099 | ||
| CNVR9 | 1 | 37,762,712–37,775,698 | Both | 5.84 × 10−05 | 0.087 | ||
| FC | CNVR10 | 14 | 51,244,826–51,249,961 | Deletion | 5.58 × 10−05 | 0.077 | CSMD3 |
| CNVR11 | 15 | 47,438,725–47,455,722 | Both | 3.10 × 10−05 | 0.077 | OR56B2C | |
| PH | CNVR12 | 7 | 40,149,305–40,156,641 | Deletion | 9.28 × 10−06 | 0.075 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Wu, T.; Xie, X.; Niu, Q.; Zhao, Z.; Zhu, B.; Chen, Y.; Zhang, L.; Gao, X.; Niu, X.; et al. Genetic Association Analysis of Copy Number Variations for Meat Quality in Beef Cattle. Foods 2023, 12, 3986. https://doi.org/10.3390/foods12213986
Wu J, Wu T, Xie X, Niu Q, Zhao Z, Zhu B, Chen Y, Zhang L, Gao X, Niu X, et al. Genetic Association Analysis of Copy Number Variations for Meat Quality in Beef Cattle. Foods. 2023; 12(21):3986. https://doi.org/10.3390/foods12213986
Chicago/Turabian StyleWu, Jiayuan, Tianyi Wu, Xueyuan Xie, Qunhao Niu, Zhida Zhao, Bo Zhu, Yan Chen, Lupei Zhang, Xue Gao, Xiaoyan Niu, and et al. 2023. "Genetic Association Analysis of Copy Number Variations for Meat Quality in Beef Cattle" Foods 12, no. 21: 3986. https://doi.org/10.3390/foods12213986