
Mechanistic Insights of Polyphenolic Compounds from Rosemary Bound to Their Protein Targets Obtained by Molecular Dynamics Simulations and Free-Energy Calculations

Abstract
:1. Introduction
2. Materials and Methods
2.1. Target Preparation and Docking
2.2. Molecular Dynamics Simulations and LIE Calculations
3. Results and Discussion
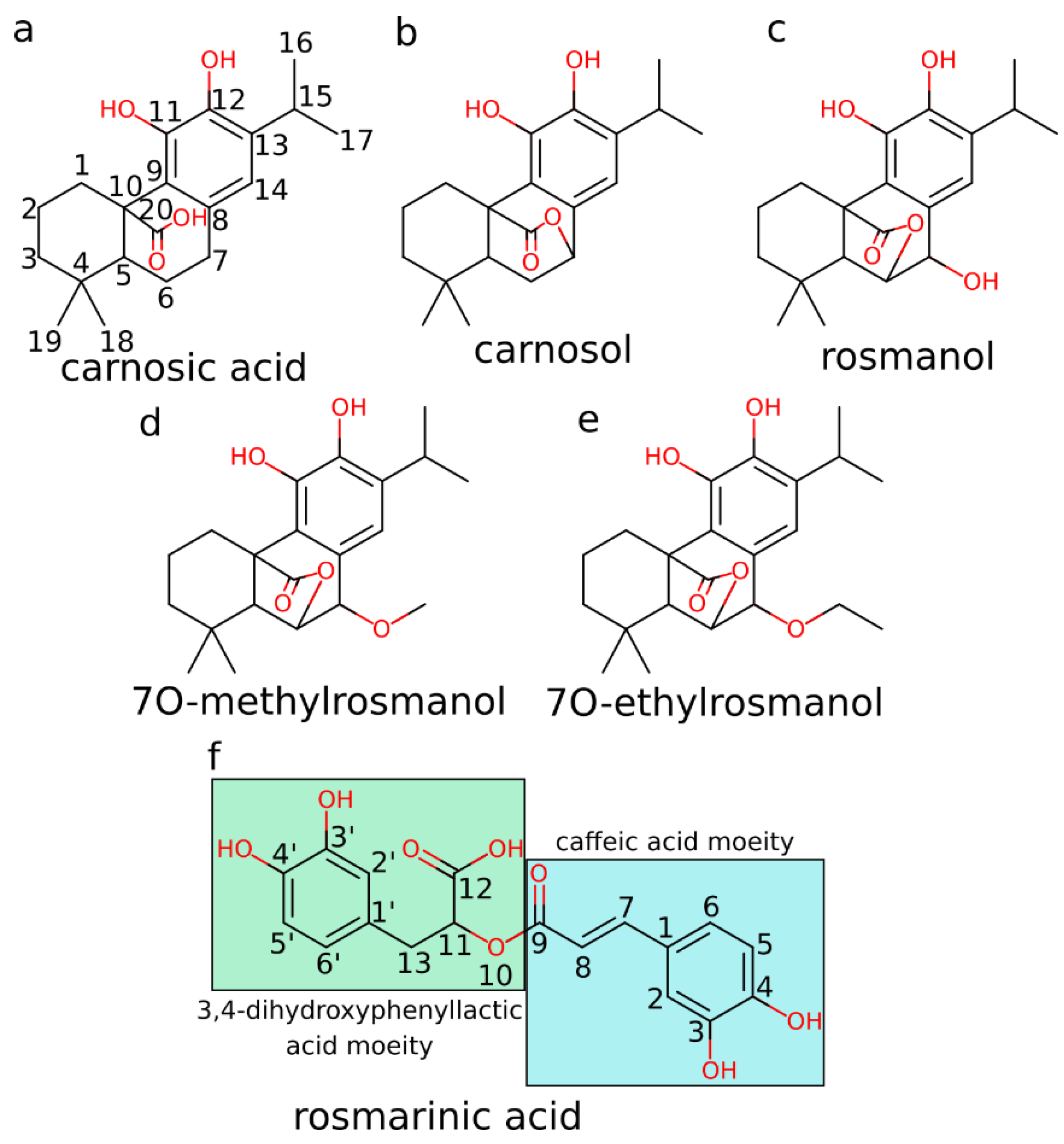
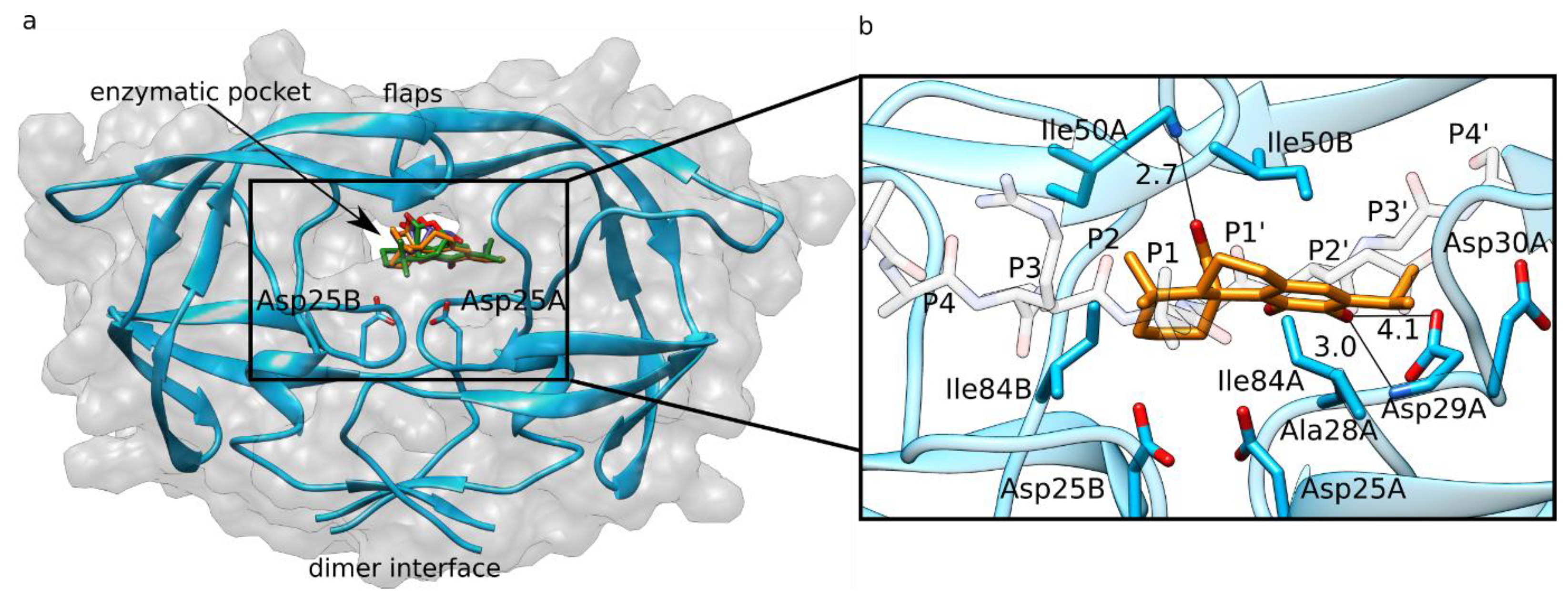
3.1. HIV-1 Protease Molecular Docking
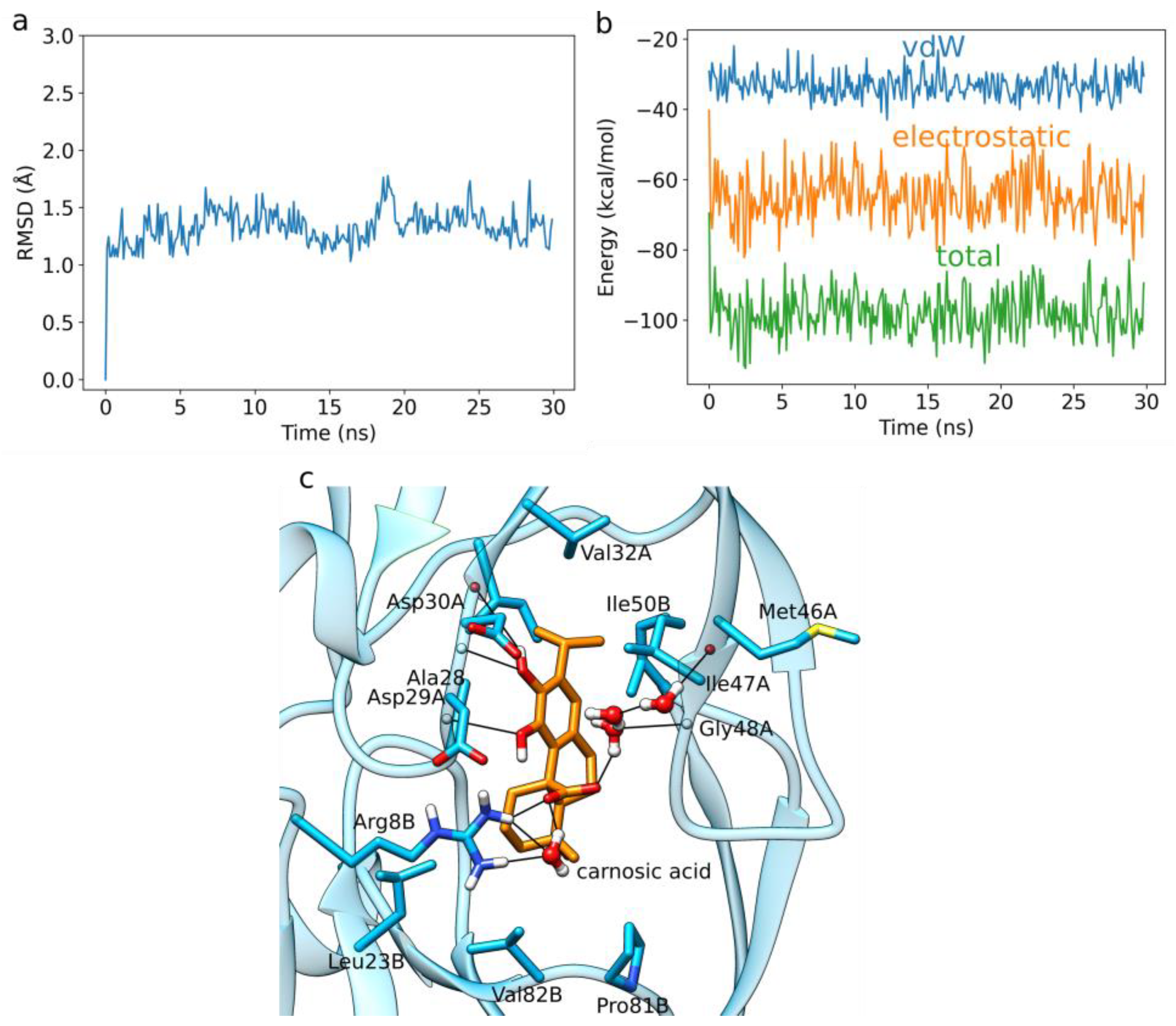
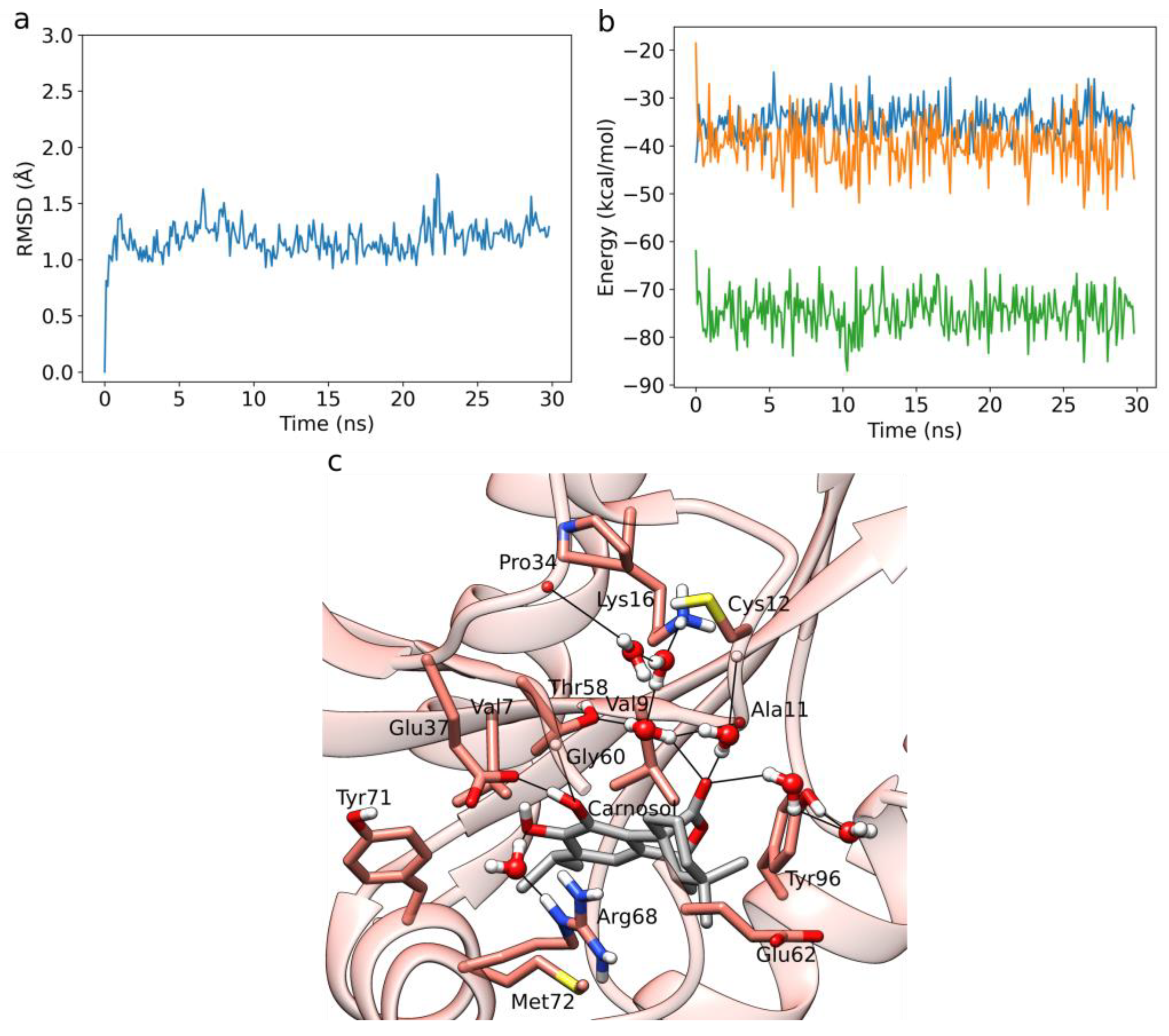
3.2. HIV-1 Protease MD Simulations and LIE Calculations
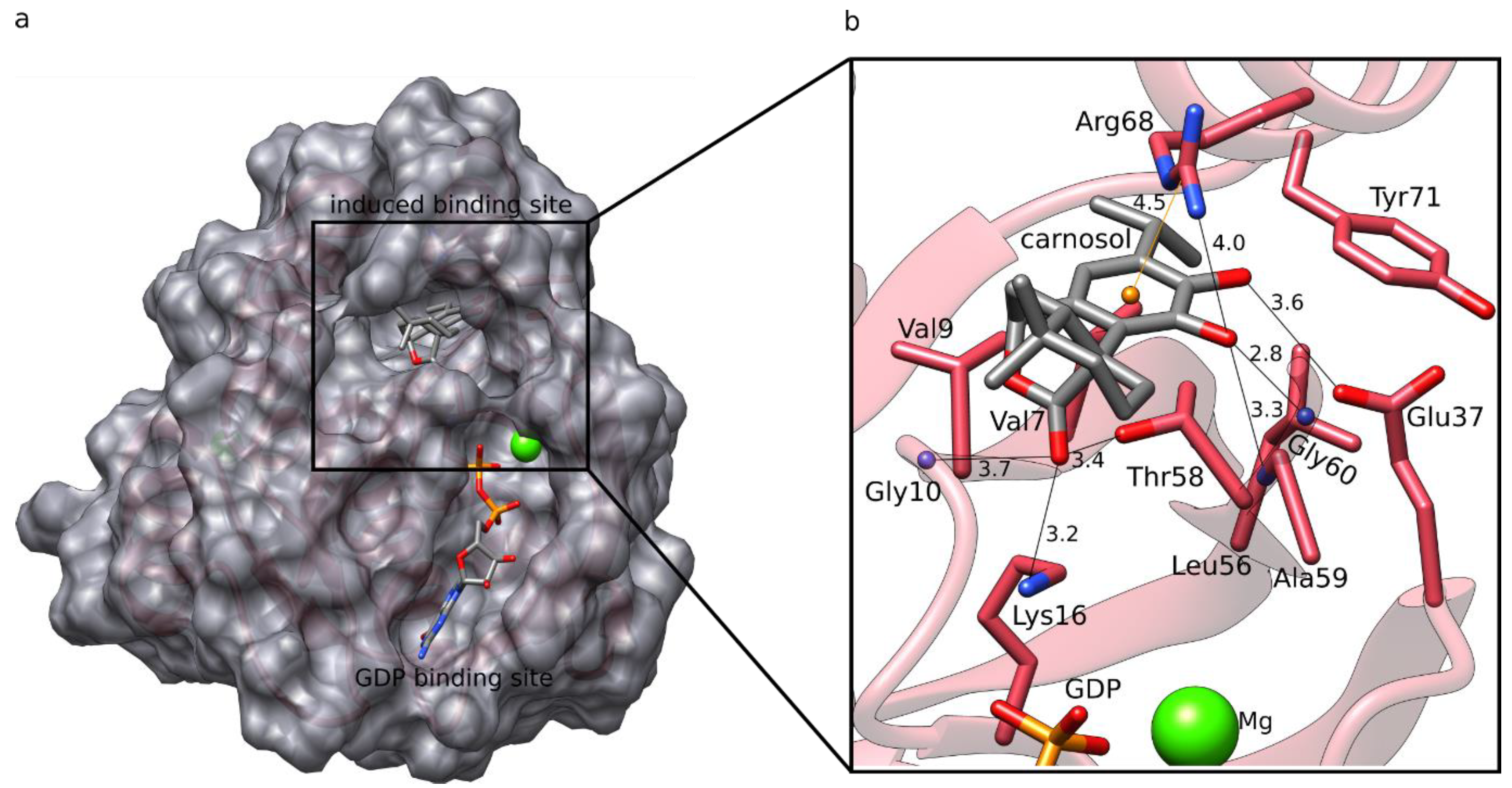
3.3. K-RAS Docking
3.4. K-RAS MD Simulations and LIE Calculations
3.5. Factor X Docking
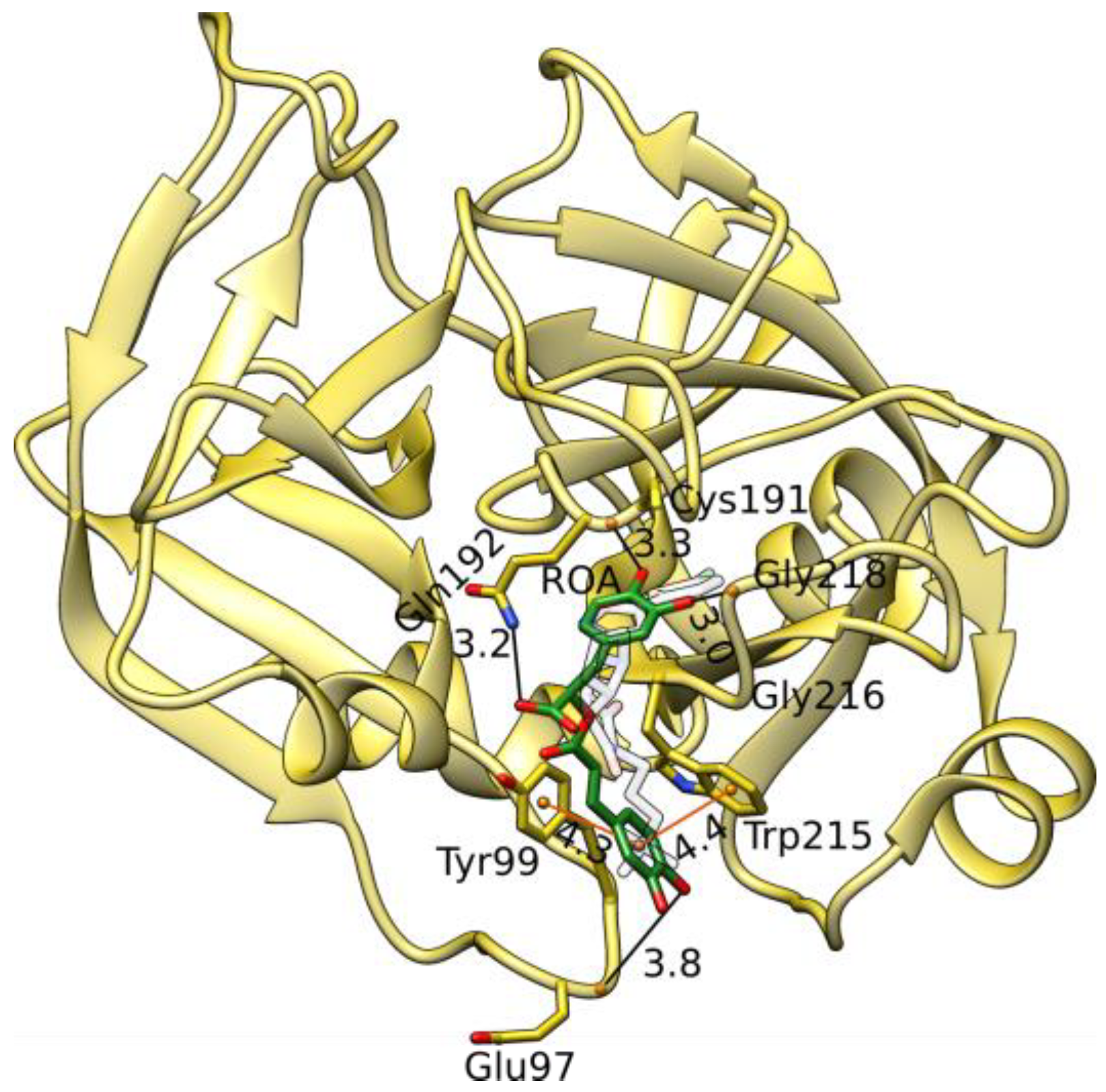
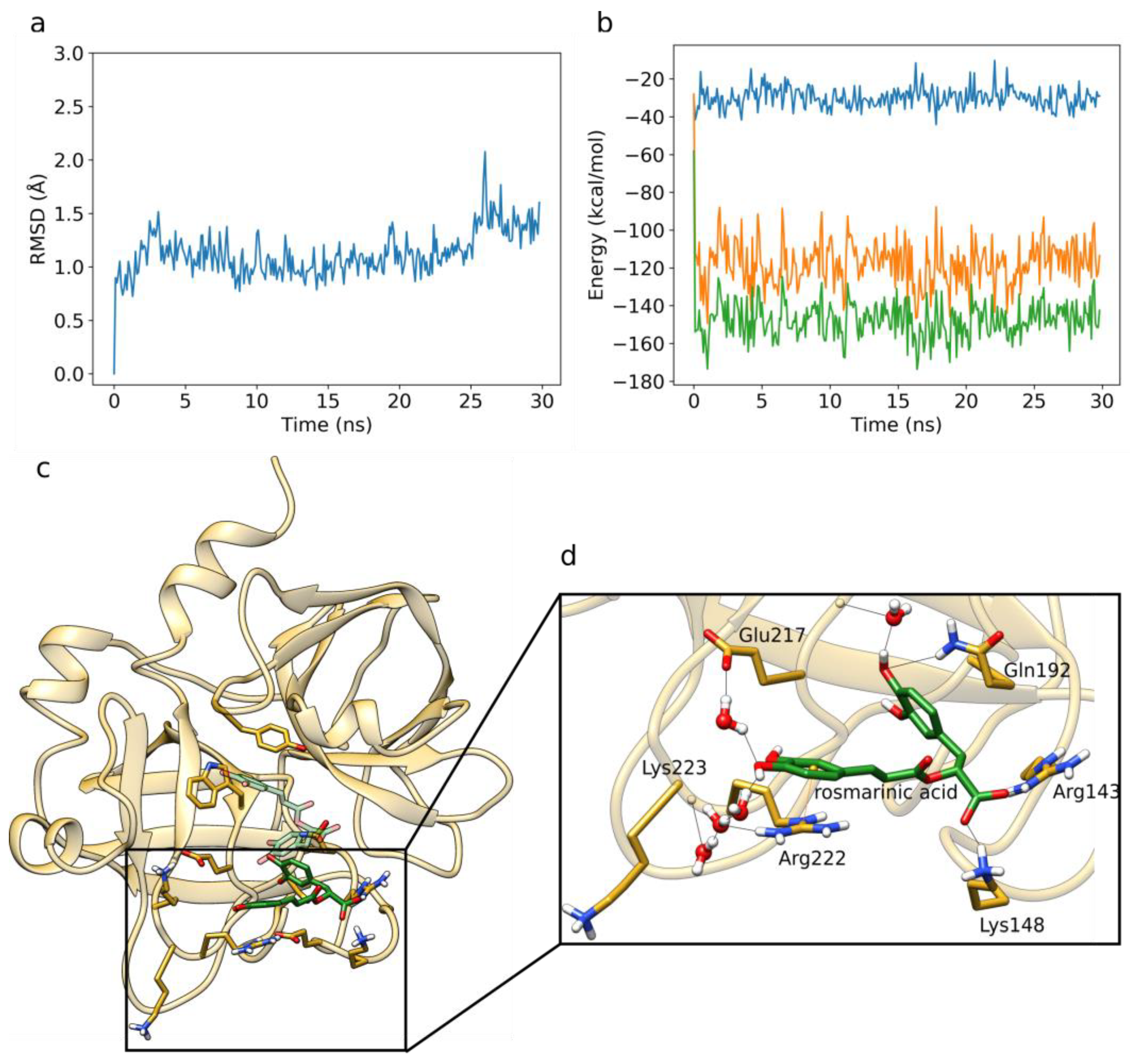
3.6. Factor X MD Simulations and LIE Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | Molecular dynamics |
| LIE | Linear interaction energy |
| HIV-1 | Human immunodeficiency virus 1 |
| K-RAS | Kirsten-rat sarcoma viral oncogene homolog |
| RAS/MAPK | Rat sarcoma/mitogen-activated protein kinase |
| GTP | Guanosine triphosphate |
| PDB | Protein Data Bank |
| CHARMM | Chemistry at Harvard Macromolecular Mechanics |
| CHARMM-GUI | Chemistry at Harvard Macromolecular Mechanics graphical user interface |
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [Green Version]
- Brglez Mojzer, E.; Knez Hrnčič, M.; Škerget, M.; Knez, Ž.; Bren, U. Polyphenols: Extraction Methods, Antioxidative Action, Bioavailability and Anticarcinogenic Effects. Molecules 2016, 21, 901. [Google Scholar] [CrossRef]
- Lešnik, S.; Furlan, V.; Bren, U. Rosemary (Rosmarinus officinalis L.): Extraction techniques, analytical methods and health-promoting biological effects. Phytochem. Rev. 2021, 20, 1273–1328. [Google Scholar] [CrossRef]
- Pérez-Fons, L.; Garzón, M.T.; Micol, V. Relationship between the Antioxidant Capacity and Effect of Rosemary (Rosmarinus officinalis L.) Polyphenols on Membrane Phospholipid Order. J. Agric. Food Chem. 2009, 58, 161–171. [Google Scholar] [CrossRef]
- Yu, M.-H.; Choi, J.-H.; Chae, I.-G.; Im, H.-G.; Yang, S.-A.; More, K.; Lee, I.-S.; Lee, J. Suppression of LPS-induced inflammatory activities by Rosmarinus officinalis L. Food Chem. 2013, 136, 1047–1054. [Google Scholar] [CrossRef]
- Bozin, B.; Mimica-Dukic, N.; Samojlik, I.; Jovin, E. Antimicrobial and Antioxidant Properties of Rosemary and Sage (Rosmarinus officinalis L. and Salvia officinalis L., Lamiaceae) Essential Oils. J. Agric. Food Chem. 2007, 55, 7879–7885. [Google Scholar] [CrossRef]
- Bakırel, T.; Bakırel, U.; Keleş, O.; Ülgen, S.G.; Yardibi, H. In vivo assessment of antidiabetic and antioxidant activities of rosemary (Rosmarinus officinalis) in alloxan-diabetic rabbits. J. Ethnopharmacol. 2008, 116, 64–73. [Google Scholar] [CrossRef]
- Hussain, S.M.; Syeda, A.F.; Alshammari, M.; Alnasser, S.; Alenzi, N.D.; Alanazi, S.T.; Nandakumar, K. Cognition enhancing effect of rosemary (Rosmarinus officinalis L.) in lab animal studies: A systematic review and meta-analysis. Braz. J. Med. Biol. Res. 2022, 55, e11593. [Google Scholar] [CrossRef]
- Moss, M.; Smith, E.; Milner, M.; McCready, J. Acute ingestion of rosemary water: Evidence of cognitive and cerebrovascular effects in healthy adults. J. Psychopharmacol. 2018, 32, 1319–1329. [Google Scholar] [CrossRef]
- Tai, J.; Cheung, S.; Wu, M.; Hasman, D. Antiproliferation effect of Rosemary (Rosmarinus officinalis) on human ovarian cancer cells in vitro. Phytomedicine 2012, 19, 436–443. [Google Scholar] [CrossRef]
- Valdés, A.; García-Cañas, V.; Rocamora-Reverte, L.; Gómez-Martínez, Á.; Ferragut, J.A.; Cifuentes, A. Effect of rosemary polyphenols on human colon cancer cells: Transcriptomic profiling and functional enrichment analysis. Genes Nutr. 2013, 8, 43–60. [Google Scholar] [CrossRef] [Green Version]
- Yesil-Celiktas, O.; Sevimli, C.; Bedir, E.; Vardar-Sukan, F. Inhibitory Effects of Rosemary Extracts, Carnosic Acid and Rosmarinic Acid on the Growth of Various Human Cancer Cell Lines. Plant Foods Hum. Nutr. 2010, 65, 158–163. [Google Scholar] [CrossRef]
- Singletary, K.; MacDonald, C.; Wallig, M. Inhibition by rosemary and carnosol of 7,12-dimethylbenz[a]anthracene (DMBA)-induced rat mammary tumorigenesis and in vivo DMBA-DNA adduct formation. Cancer Lett. 1996, 104, 43–48. [Google Scholar] [CrossRef]
- Huang, M.T.; Ho, C.T.; Wang, Z.Y.; Ferraro, T.; Lou, Y.R.; Stauber, K.; Ma, W.; Georgiadis, C.; Laskin, J.D.; Conney, A.H. Inhibition of skin tumorigenesis by rosemary and its constituents carnosol and ursolic acid. Cancer Res. 1994, 54, 701–708. [Google Scholar]
- Okamura, N.; Fujimoto, Y.; Kuwabara, S.; Yagi, A. High-performance liquid chromatographic determination of carnosic acid and carnosol in Rosmarinus officinalis and Salvia officinalis. J. Chromatogr. A 1994, 679, 381–386. [Google Scholar] [CrossRef]
- Amoah, S.K.S.; Sandjo, L.P.; Kratz, J.M.; Biavatti, M.W. Rosmarinic Acid–Pharmaceutical and Clinical Aspects. Planta Med. 2016, 82, 388–406. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Provan, G.J.; Helliwell, K. Determination of rosmarinic acid and caffeic acid in aromatic herbs by HPLC. Food Chem. 2004, 87, 307–311. [Google Scholar] [CrossRef]
- Lešnik, S.; Bren, U. Mechanistic Insights into Biological Activities of Polyphenolic Compounds from Rosemary Obtained by Inverse Molecular Docking. Foods 2021, 11, 67. [Google Scholar] [CrossRef]
- Weichseldorfer, M.; Reitz, M.; Latinovic, O.S. Past HIV-1 Medications and the Current Status of Combined Antiretroviral Therapy Options for HIV-1 Patients. Pharmaceutics 2021, 13, 1798. [Google Scholar] [CrossRef]
- Kerk, S.A.; Papagiannakopoulos, T.; Shah, Y.M.; Lyssiotis, C.A. Metabolic networks in mutant KRAS-driven tumours: Tissue specificities and the microenvironment. Nat. Rev. Cancer 2021, 21, 510–525. [Google Scholar] [CrossRef]
- Ahmad, H.H.; Hamza, A.H.; Hassan, A.Z.; Sayed, A.H. Promising Therapeutic Role of Rosmarinus Officinalis Successive Methanolic Fraction against Colorectal Cancer. Int. J. Pharm. Pharm. Sci. 2013, 5, 164–170. [Google Scholar]
- Chen, X.; Zhou, L.; Zhang, Y.; Yi, D.; Liu, L.; Rao, W.; Wu, Y.; Ma, D.; Liu, X.; Zhou, X.-H.A.; et al. Risk Factors of Stroke in Western and Asian Countries: A Systematic Review and Meta-analysis of Prospective Cohort Studies. BMC Public Health 2014, 14, 776. [Google Scholar] [CrossRef]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS Auckl. 2015, 7, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Pariš, A.; Štrukelj, B.; Renko, M.; Turk, V.; Pukl, M.; Umek, A.; Korant, B.D. Inhibitory Effect of Carnosolic Acid on HIV-1 Protease in Cell-Free Assays. J. Nat. Prod. 1993, 56, 1426–1430. [Google Scholar] [CrossRef]
- Yu, Y.X.; Wang, W.; Sun, H.B.; Zhang, L.L.; Wang, L.F.; Yin, Y.Y. Decoding drug resistant mechanism of V32I, I50V and I84V mutations of HIV-1 protease on amprenavir binding by using molecular dynamics simulations and MM-GBSA calculations. SAR QSAR Environ. Res. 2022, 33, 805–831. [Google Scholar] [CrossRef]
- Matikas, A.; Mistriotis, D.; Georgoulias, V.; Kotsakis, A. Targeting KRAS mutated non-small cell lung cancer: A history of failures and a future of hope for a diverse entity. Crit. Rev. Oncol. Hematol. 2017, 110, 1–12. [Google Scholar] [CrossRef]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. [Google Scholar] [CrossRef] [Green Version]
- Issahaku, A.R.; Aljoundi, A.; Soliman, M.E. Establishing the mutational effect on the binding susceptibility of AMG510 to KRAS switch II binding pocket: Computational insights. Inform. Med. Unlocked 2022, 30, 100952. [Google Scholar] [CrossRef]
- Hoffman, M.; Monroe, D.; Oliver, J.; Roberts, H. Factors IXa and Xa play distinct roles in tissue factor-dependent initiation of coagulation. Blood 1995, 86, 1794–1801. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Peng, Y.; Wang, X.; Qian, Y.; Xiang, P.; Wade, S.W.; Guo, H.; Lopez, J.A.G.; Herzog, C.A.; Handelsman, Y. Cardiovascular Events and Death after Myocardial Infarction or Ischemic Stroke in an Older Medicare Population. Clin. Cardiol. 2019, 42, 391–399. [Google Scholar] [CrossRef]
- Perzborn, E.; Roehrig, S.; Straub, A.; Kubitza, D.; Mueck, W.; Laux, V. Rivaroxaban: A New Oral Factor Xa Inhibitor. Arter. Thromb. Vasc. Biol. 2010, 30, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Jukič, M.; Škrlj, B.; Tomšič, G.; Pleško, S.; Podlipnik, Č.; Bren, U. Prioritisation of Compounds for 3CLpro Inhibitor Development on SARS-CoV-2 Variants. Molecules 2021, 26, 3003. [Google Scholar] [CrossRef]
- Carmona, S.R.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [Green Version]
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.M.; Mortenson, P.N.; Murray, C.W. Diverse, High-Quality Test Set for the Validation of Protein−Ligand Docking Performance. J. Med. Chem. 2007, 50, 726–741. [Google Scholar] [CrossRef]
- Morley, S.D.; Afshar, M. Validation of an empirical RNA-ligand scoring function for fast flexible docking using RiboDock®. J. Comput. Aided Mol. Des. 2004, 18, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Soler, D.; Westermaier, Y.; Soliva, R. Extensive benchmark of rDock as a peptide-protein docking tool. J. Comput. Aided Mol. Des. 2019, 33, 613–626. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Mackerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Khan, H.M.; MacKerell, A.D.; Reuter, N. Cation-π Interactions between Methylated Ammonium Groups and Tryptophan in the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2018, 15, 7–12. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbová, M.; Šponer, J.; Otyepka, M.; Jurečka, P.; Cheatham, T.E. Assessing the Current State of Amber Force Field Modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef] [PubMed]
- Zgarbová, M.; Luque, F.J.; Sponer, J.; Cheatham, T.E., III; Otyepka, M.; Jurečka, P. Toward Improved Description of DNA Backbone: Revisiting Epsilon and Zeta Torsion Force Field Parameters. J. Chem. Theory Comput. 2013, 9, 2339–2354. [Google Scholar] [CrossRef] [PubMed]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; Perera, L.; Li, L.; Pedersen, L. New tricks for modelers from the crystallography toolkit: The particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure 1999, 7, R55–R60. [Google Scholar] [CrossRef] [Green Version]
- Åqvist, J.; Medina, C.; Samuelsson, J.-E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. Des. Sel. 1994, 7, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Hansson, T.; Marelius, J.; Åqvist, J. Ligand binding affinity prediction by linear interaction energy methods. J. Comput. Aided Mol. Des. 1998, 12, 27–35. [Google Scholar] [CrossRef]
- van Dijk, M.; ter Laak, A.M.; Wichard, J.D.; Capoferri, L.; Vermeulen, N.P.E.; Geerke, D.P. Comprehensive and Automated Linear Interaction Energy Based Binding-Affinity Prediction for Multifarious Cytochrome P450 Aromatase Inhibitors. J. Chem. Inf. Model. 2017, 57, 2294–2308. [Google Scholar] [CrossRef] [Green Version]
- Rifai, E.A.; van Dijk, M.; Vermeulen, N.P.E.; Yanuar, A.; Geerke, D.P. A Comparative Linear Interaction Energy and MM/PBSA Study on SIRT1–Ligand Binding Free Energy Calculation. J. Chem. Inf. Model. 2019, 59, 4018–4033. [Google Scholar] [CrossRef] [Green Version]
- Rahaman, O.; Estrada, T.P.; Doren, D.J.; Taufer, M.; Brooks, I.C.L.; Armen, R.S. Evaluation of Several Two-Step Scoring Functions Based on Linear Interaction Energy, Effective Ligand Size, and Empirical Pair Potentials for Prediction of Protein–Ligand Binding Geometry and Free Energy. J. Chem. Inf. Model. 2011, 51, 2047–2065. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Caflisch, A. Efficient Evaluation of Binding Free Energy Using Continuum Electrostatics Solvation. J. Med. Chem. 2004, 47, 5791–5797. [Google Scholar] [CrossRef]
- Singh, P.; Mhaka, A.M.; Christensen, S.B.; Gray, J.J.; Denmeade, S.R.; Isaacs, J.T. Applying Linear Interaction Energy Method for Rational Design of Noncompetitive Allosteric Inhibitors of the Sarco- and Endoplasmic Reticulum Calcium-ATPase. J. Med. Chem. 2005, 48, 3005–3014. [Google Scholar] [CrossRef]
- Almlöf, M.; Brandsdal, B.O.; Åqvist, J. Binding affinity prediction with different force fields: Examination of the linear interaction energy method. J. Comput. Chem. 2004, 25, 1242–1254. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Mai, B.K.; Derreumaux, P.; Vu, V.V. Adequate prediction for inhibitor affinity of Aβ40 protofibril using the linear interaction energy method. RSC Adv. 2019, 9, 12455–12461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, E.; Oliphant, T.; Peterson, P. SciPy: Open Source Scientific Tools for Python. 2001. Available online: http://www.scipy.org (accessed on 5 November 2022).
- Ngo, S.T.; Hong, N.D.; Anh, L.H.Q.; Hiep, D.M.; Tung, N.T. Effective estimation of the inhibitor affinity of HIV-1 protease via a modified LIE approach. RSC Adv. 2020, 10, 7732–7739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, M.; Bouguet-Bonnet, S.; Pastor, R.W.; MacKerell, A.D. Importance of the CMAP Correction to the CHARMM22 Protein Force Field: Dynamics of Hen Lysozyme. Biophys. J. 2006, 90, L36–L38. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Peck, J.M.; Ilgu, M.; Nilsen-Hamilton, M.; Lamm, M.H. Sampling Performance of Multiple Independent Molecular Dynamics Simulations of an RNA Aptamer. ACS Omega 2020, 5, 20187–20201. [Google Scholar] [CrossRef]
- Grossfield, A.; Patrone, P.N.; Roe, D.R.; Schultz, A.J.; Siderius, D.W.; Zuckerman, D.M. Best Practices for Quantification of Uncertainty and Sampling Quality in Molecular Simulations [Article v1.0]. Living J. Comput. Mol. Sci. 2018, 1, 5067. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.J.; Tomas, M.S.; Rubio-Martinez, J. Assessment of the Sampling Performance of Multiple-Copy Dynamics versus a Unique Trajectory. J. Chem. Inf. Model. 2016, 56, 1950–1962. [Google Scholar] [CrossRef]
- Stjernschantz, E.; Oostenbrink, C. Improved Ligand-Protein Binding Affinity Predictions Using Multiple Binding Modes. Biophys. J. 2010, 98, 2682–2691. [Google Scholar] [CrossRef] [Green Version]
- Klvana, M.; Bren, U. Aflatoxin B1–Formamidopyrimidine DNA Adducts: Relationships between Structures, Free Energies, and Melting Temperatures. Molecules 2019, 24, 150. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domanski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for The Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016; Volume 98, p. 105. [Google Scholar]
- Siemers, M.; Lazaratos, M.; Karathanou, K.; Guerra, F.; Brown, L.S.; Bondar, A.-N. Bridge: A Graph-Based Algorithm to Analyze Dynamic H-Bond Networks in Membrane Proteins. J. Chem. Theory Comput. 2019, 15, 6781–6798. [Google Scholar] [CrossRef]
- Siemers, M.; Bondar, A.-N. Interactive Interface for Graph-Based Analyses of Dynamic H-Bond Networks: Application to Spike Protein S. J. Chem. Inf. Model. 2021, 61, 2998–3014. [Google Scholar] [CrossRef] [PubMed]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.; Malito, E.; Shen, Y.; Pentelute, B.; Rich, D.; Florián, J.; Tang, W.-J.; Kent, S.B. Insights from Atomic-Resolution X-Ray Structures of Chemically Synthesized HIV-1 Protease in Complex with Inhibitors. J. Mol. Biol. 2007, 373, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Inaba, Y.; Maekawa, T.; Takeda, Y.; Tamura, H.; Yamaguchi, H. Recovery Mechanism of the Antioxidant Activity from Carnosic Acid Quinone, an Oxidized Sage and Rosemary Antioxidant. J. Agric. Food Chem. 2002, 50, 5863–5869. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yan, H.; Chen, J.; Wang, Y.; Jiang, Y.; Tu, P. Characterization of in vitro and in vivo metabolites of carnosic acid, a natural antioxidant, by high performance liquid chromatography coupled with tandem mass spectrometry. J. Pharm. Biomed. Anal. 2014, 89, 183–196. [Google Scholar] [CrossRef]
- Vaquero, M.R.; Villalba, R.G.; Larrosa, M.; Yáñez-Gascón, M.J.; Fromentin, E.; Flanagan, J.; Roller, M.; Tomás-Barberán, F.A.; Espín, J.C.; García-Conesa, M.-T. Bioavailability of the major bioactive diterpenoids in a rosemary extract: Metabolic profile in the intestine, liver, plasma, and brain of Zucker rats. Mol. Nutr. Food Res. 2013, 57, 1834–1846. [Google Scholar] [CrossRef]
- Doolaege, E.H.A.; Raes, K.; De Vos, F.; Verhé, R.; De Smet, S. Absorption, Distribution and Elimination of Carnosic Acid, A Natural Antioxidant from Rosmarinus officinalis, in Rats. Plant Foods Hum. Nutr. 2011, 66, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Vemu, B.; Tocmo, R.; Nauman, M.C.; Flowers, S.A.; Veenstra, J.P.; Johnson, J.J. Pharmacokinetic characterization of carnosol from rosemary (Salvia Rosmarinus) in male C57BL/6 mice and inhibition profile in human cytochrome P450 enzymes. Toxicol. Appl. Pharmacol. 2021, 431, 115729. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gan, C.; Wang, Z.; Liu, L.; Gao, M.; Li, Q.; Yang, C. Determination and Pharmacokinetic Study of Three Diterpenes in Rat Plasma by UHPLC-ESI-MS/MS after Oral Administration of Rosmarinus officinalis L. Extract. Mol. 2017, 22, 934. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput. Aided Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef]
- Maurer, M.; Oostenbrink, C. Water in protein hydration and ligand recognition. J. Mol. Recognit. 2019, 32, e2810. [Google Scholar] [CrossRef] [PubMed]
- Hitl, M.; Kladar, N.; Gavarić, N.; Božin, B. Rosmarinic Acid–Human Pharmacokinetics and Health Benefits. Planta Med. 2021, 87, 273–282. [Google Scholar] [CrossRef]
- Nakazawa, T.; Ohsawa, K. Metabolism of Rosmarinic Acid in Rats. J. Nat. Prod. 1998, 61, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Baba, S.; Osakabe, N.; Natsume, M.; Yasuda, A.; Muto, Y.; Hiyoshi, K.; Takano, H.; Yoshikawa, T.; Terao, J. Absorption, metabolism, degradation and urinary excretion of rosmarinic acid after intake of Perilla frutescens extract in humans. Eur. J. Nutr. 2005, 44, 1–9. [Google Scholar] [CrossRef]
- Wang, J.; Li, G.; Rui, T.; Kang, A.; Li, G.; Fu, T.; Li, J.; Di, L.; Cai, B. Pharmacokinetics of rosmarinic acid in rats by LC-MS/MS: Absolute bioavailability and dose proportionality. RSC Adv. 2017, 7, 9057–9063. [Google Scholar] [CrossRef] [Green Version]
- Achour, M.; Saguem, S.; Sarriá, B.; Bravo, L.; Mateos, R. Bioavailability and metabolism of rosemary infusion polyphenols using Caco-2 and HepG2 cell model systems. J. Sci. Food Agric. 2018, 98, 3741–3751. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wen, Q.; Qian, K.; Feng, Y.; Luo, Y.; Tan, T. Metabolic profile of rosmarinic acid from Java tea (Orthosiphon stamineus) by ultra-high-performance liquid chromatography coupled to quadrupole-time-of-flight tandem mass spectrometry with a three-step data mining strategy. Biomed. Chromatogr. 2019, 33, e4599. [Google Scholar] [CrossRef]
- Su, J.; Jia, F.; Lu, J.; Chen, W.; Sun, H.; Liu, T.; Wu, X. Characterization of the metabolites of rosmarinic acid in human liver microsomes using liquid chromatography combined with electrospray ionization tandem mass spectrometry. Biomed. Chromatogr. 2020, 34, e4806. [Google Scholar] [CrossRef] [PubMed]
- Noguchi-Shinohara, M.; Ono, K.; Hamaguchi, T.; Iwasa, K.; Nagai, T.; Kobayashi, S.; Nakamura, H.; Yamada, M. Pharmacokinetics, Safety and Tolerability of Melissa officinalis Extract which Contained Rosmarinic Acid in Healthy Individuals: A Randomized Controlled Trial. PLoS ONE 2015, 10, e0126422. [Google Scholar] [CrossRef] [PubMed]
- Florio, P.; Folli, C.; Cianci, M.; Del Rio, D.; Zanotti, G.; Berni, R. Transthyretin Binding Heterogeneity and Anti-amyloidogenic Activity of Natural Polyphenols and Their Metabolites. J. Biol. Chem. 2015, 290, 29769–29780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvador, G.H.M.; Cardoso, F.F.; Gomes, A.A.; Cavalcante, W.L.G.; Gallacci, M.; Fontes, M.R.M. Search for efficient inhibitors of myotoxic activity induced by ophidian phospholipase A2-like proteins using functional, structural and bioinformatics approaches. Sci. Rep. 2019, 9, 510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzei, L.; Cianci, M.; Musiani, F.; Lente, G.; Palombo, M.; Ciurli, S. Inactivation of urease by catechol: Kinetics and structure. J. Inorg. Biochem. 2017, 166, 182–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, G. The role of polyphenols in modern nutrition. Nutr. Bull. 2017, 42, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Pimpão, R.C.; Ventura, M.R.; Ferreira, R.B.; Williamson, G.; Santos, C.N. Phenolic sulfates as new and highly abundant metabolites in human plasma after ingestion of a mixed berry fruit purée. Br. J. Nutr. 2015, 113, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Monagas, M.; Urpi-Sarda, M.; Sánchez-Patán, F.; Llorach, R.; Garrido, I.; Gómez-Cordovés, C.; Andres-Lacueva, C.; Bartolomé, B. Insights into the metabolism and microbial biotransformation of dietary flavan-3-ols and the bioactivity of their metabolites. Food Funct. 2010, 1, 233–253. [Google Scholar] [CrossRef] [Green Version]
- Gakh, A.A.; Anisimova, N.Y.; Kiselevsky, M.V.; Sadovnikov, S.V.; Stankov, I.N.; Yudin, M.V.; Rufanov, K.A.; Krasavin, M.Y.; Sosnov, A.V. Dihydro-resveratrol—A potent dietary polyphenol. Bioorg. Med. Chem. Lett. 2010, 20, 6149–6151. [Google Scholar] [CrossRef]
- Derbyshire, D.J.; Hammarstrom, P.; von Castelmur, E.; Begum, A. Transthyretin Binding Mode Ambivalence of Fluorescent Trans-Stilbene Probes. to be published.
- Chopra, H.; Bibi, S.; Islam, F.; Ahmad, S.U.; Olawale, O.A.; Alhumaydhi, F.A.; Marzouki, R.; Baig, A.A.; Bin Emran, T. Emerging Trends in the Delivery of Resveratrol by Nanostructures: Applications of Nanotechnology in Life Sciences. J. Nanomater. 2022, 2022, 3083728. [Google Scholar] [CrossRef]
- Ge, Z.-Q.; Du, X.-Y.; Huang, X.-N.; Qiao, B. Enhanced oral bioavailability of ursolic acid nanoparticles via antisolvent precipitation with TPGS1000 as a stabilizer. J. Drug Deliv. Sci. Technol. 2015, 29, 210–217. [Google Scholar] [CrossRef]
- Yang, L.; Sun, Z.; Zu, Y.; Zhao, C.; Sun, X.; Zhang, Z.; Zhang, L. Physicochemical properties and oral bioavailability of ursolic acid nanoparticles using supercritical anti-solvent (SAS) process. Food Chem. 2012, 132, 319–325. [Google Scholar] [CrossRef]
- Bhise, K.; Kashaw, S.K.; Sau, S.; Iyer, A.K. Nanostructured lipid carriers employing polyphenols as promising anticancer agents: Quality by design (QbD) approach. Int. J. Pharm. 2017, 526, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Zupančič, Š.; Kocbek, P.; Zariwala, M.G.; Renshaw, D.; Gul, M.O.; Elsaid, Z.; Taylor, K.M.G.; Somavarapu, S. Design and Development of Novel Mitochondrial Targeted Nanocarriers, DQAsomes for Curcumin Inhalation. Mol. Pharm. 2014, 11, 2334–2345. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand/Parameter Set | vdW Contribution (kcal/mol) | Coulomb Contribution (kcal/mol) | ΔGBIND (kcal/mol) |
|---|---|---|---|
| Carnosic acid | |||
| α = 0.76, β = 0.24 | −7.35 ± 0.63 | −1.22 ± 0.17 | −8.56 ± 0.76 |
| Carnosol | |||
| α = 0.76, β = 0.24 | −6.66 ± 1.76 | −0.18 ± 1.87 | −6.84 ± 0.41 |
| Rosmanol | |||
| α = 0.76, β = 0.24 | −6.08 ± 1.74 | −1.93 ± 1.76 | −8.01 ± 1.01 |
| 7O-methylrosmanol | |||
| α = 0.76, β = 0.24 | −4.84 ± 1.56 | −2.97 ± 1.95 | −7.81 ± 0.75 |
| 7O-ethylrosmanol | |||
| α = 0.76, β = 0.24 | −3.55 ± 0.14 | −3.89 ± 0.23 | −7.45 ± 0.12 |
| System/Parameter Set | vdW Contribution (kcal/mol) | Coulomb Contribution (kcal/mol) | ΔGBIND (kcal/mol) |
|---|---|---|---|
| Carnosic acid | |||
| (a) α = 0.18, β = 0.50 | −2.16 ± 0.24 | 2.32 ± 0.59 | 0.16 ± 0.41 |
| Carnosol | |||
| (a) α = 0.18, β = 0.33 | −1.80 ± 0.09 | −4.56 ± 0.20 | −6.36 ± 0.15 |
| Rosmanol | |||
| (a) α = 0.18, β = 0.33 | −2.47 ± 0.68 | −0.50 ± 3.04 | −2.96 ± 2.36 |
| System/Set | VdW Contribution (kcal/mol) | Coulomb Contribution (kcal/mol) | ΔGBIND (kcal/mol) |
|---|---|---|---|
| Rosmarinic acid | |||
| (a) α = 0.18, β = 0.50 | −2.35 ± 0.22 | 1.83 ± 0.84 | −0.52 ± 0.64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lešnik, S.; Jukič, M.; Bren, U. Mechanistic Insights of Polyphenolic Compounds from Rosemary Bound to Their Protein Targets Obtained by Molecular Dynamics Simulations and Free-Energy Calculations. Foods 2023, 12, 408. https://doi.org/10.3390/foods12020408
Lešnik S, Jukič M, Bren U. Mechanistic Insights of Polyphenolic Compounds from Rosemary Bound to Their Protein Targets Obtained by Molecular Dynamics Simulations and Free-Energy Calculations. Foods. 2023; 12(2):408. https://doi.org/10.3390/foods12020408
Chicago/Turabian StyleLešnik, Samo, Marko Jukič, and Urban Bren. 2023. "Mechanistic Insights of Polyphenolic Compounds from Rosemary Bound to Their Protein Targets Obtained by Molecular Dynamics Simulations and Free-Energy Calculations" Foods 12, no. 2: 408. https://doi.org/10.3390/foods12020408