
Evaluation of Prebiotics through an In Vitro Gastrointestinal Digestion and Fecal Fermentation Experiment: Further Idea on the Implementation of Machine Learning Technique


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
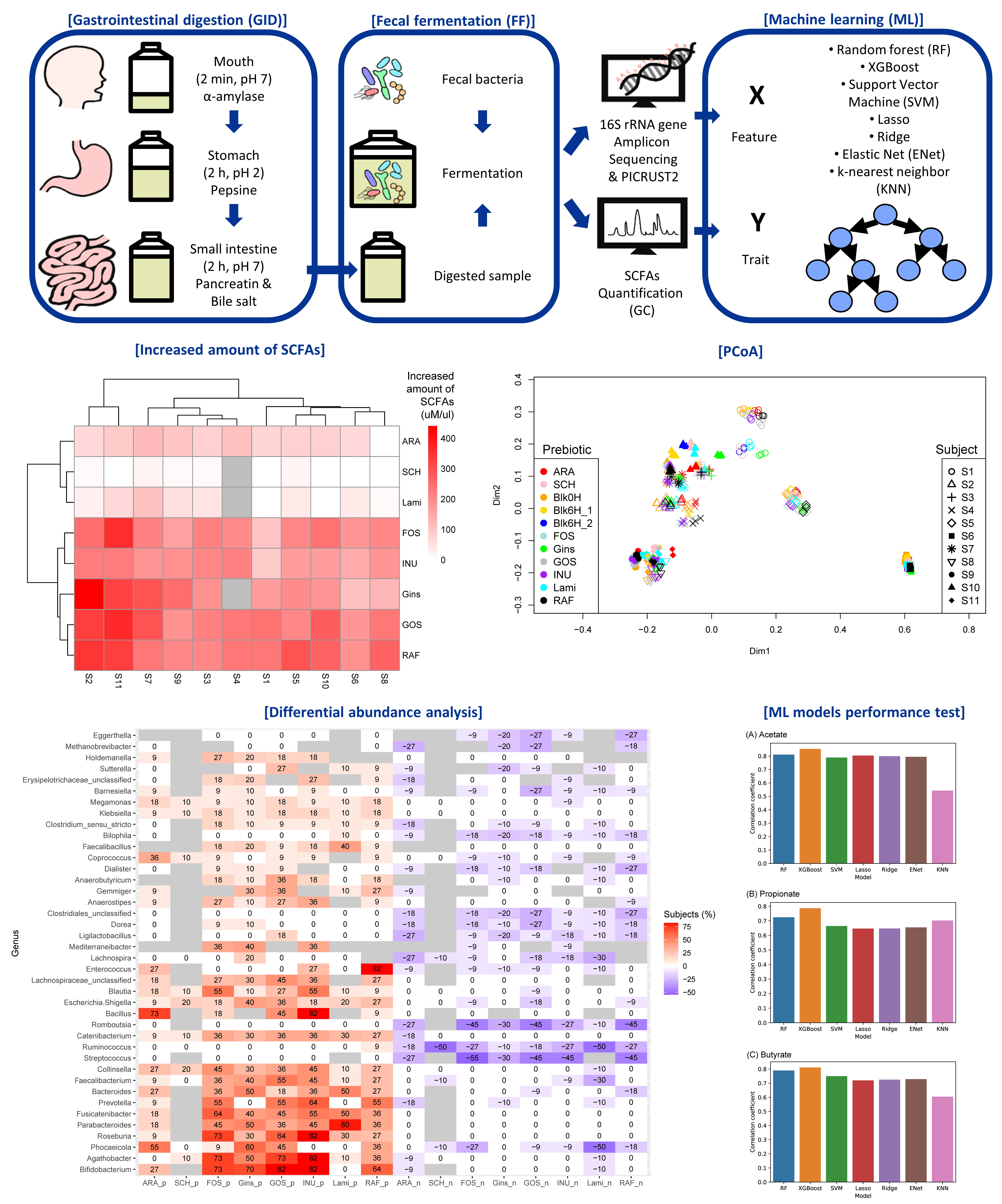
2.1. In Vitro Gastrointestinal Digestion and Fecal Fermentation
2.2. Quantification of SCFAs
2.3. Bioinformatics
2.4. Statistical Analysis
2.5. Machine Learning
3. Results and Discussion
3.1. SCFA Production after Prebiotics Treatments
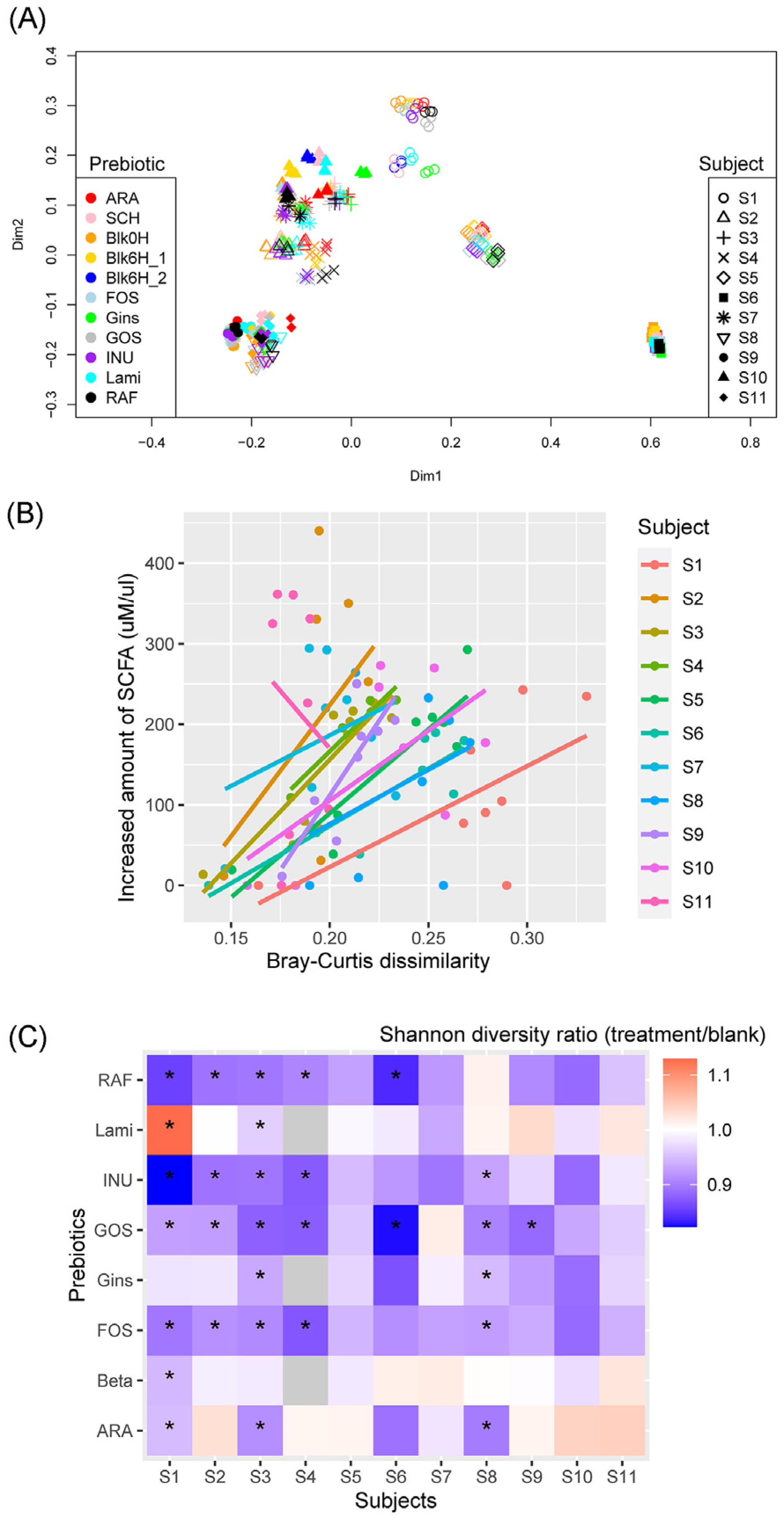
3.2. SCFA Production and Fecal Microbiota of Each Subject
3.3. Microbiota Shifts after Prebiotics Treatment
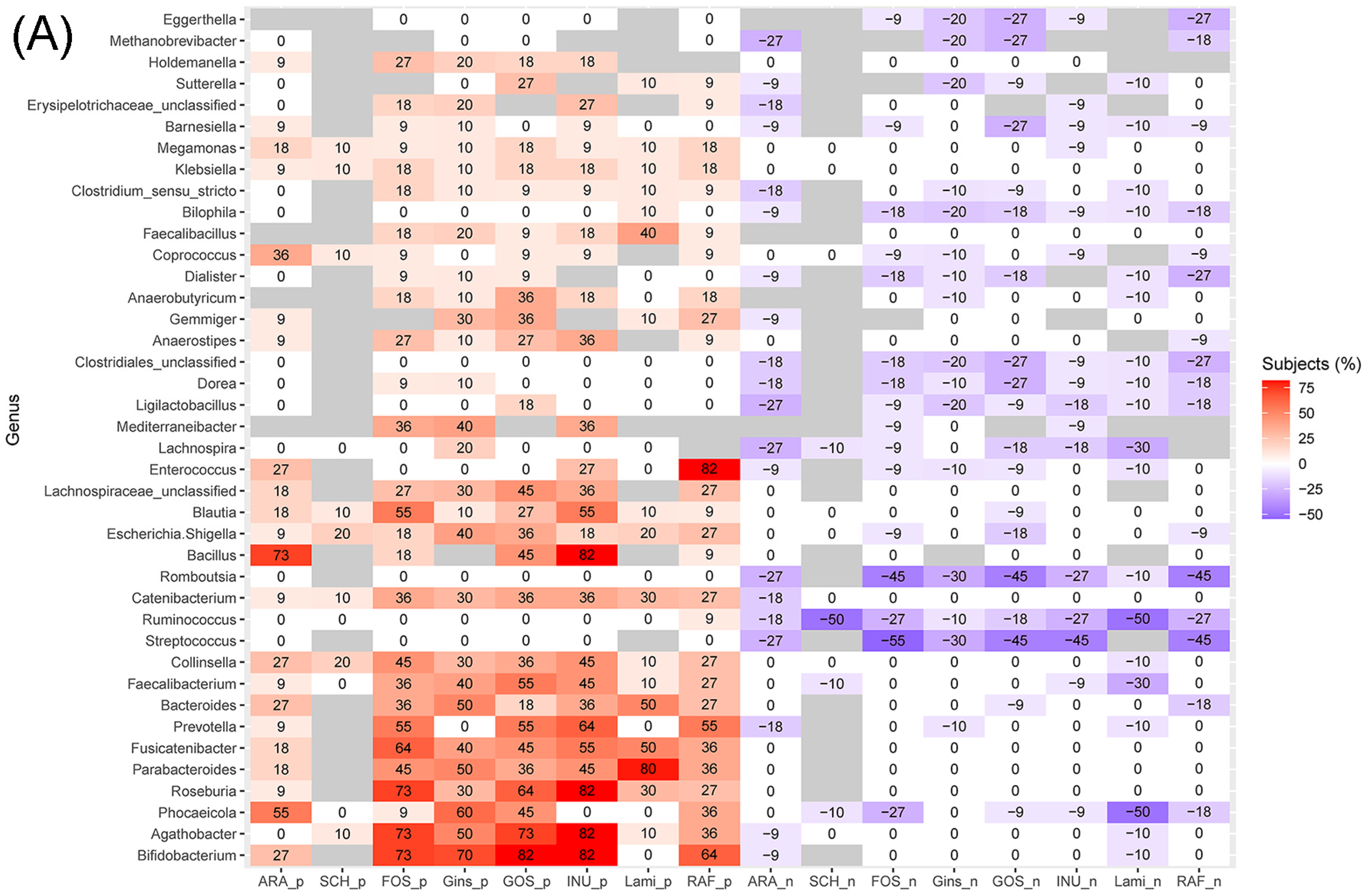
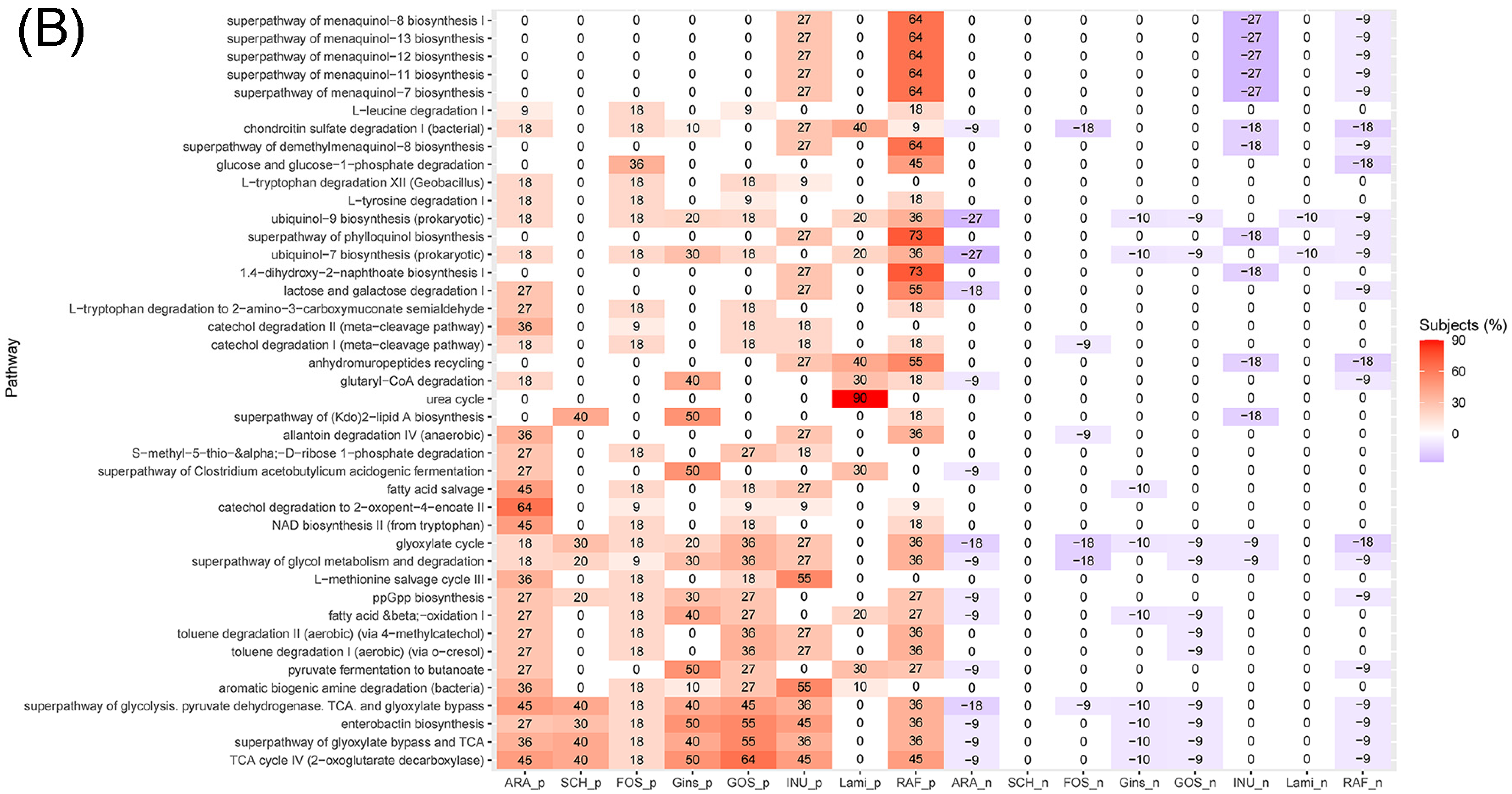
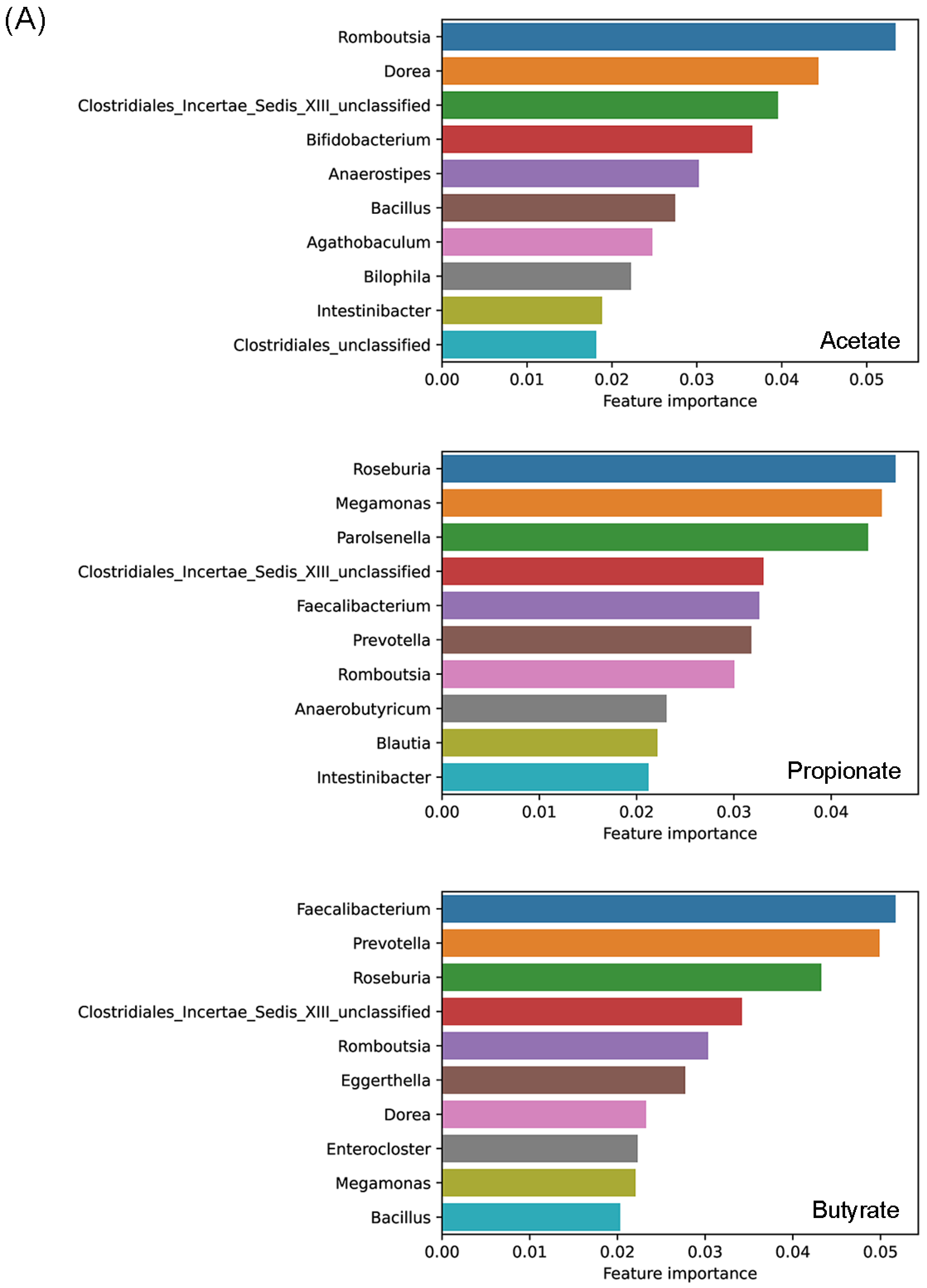
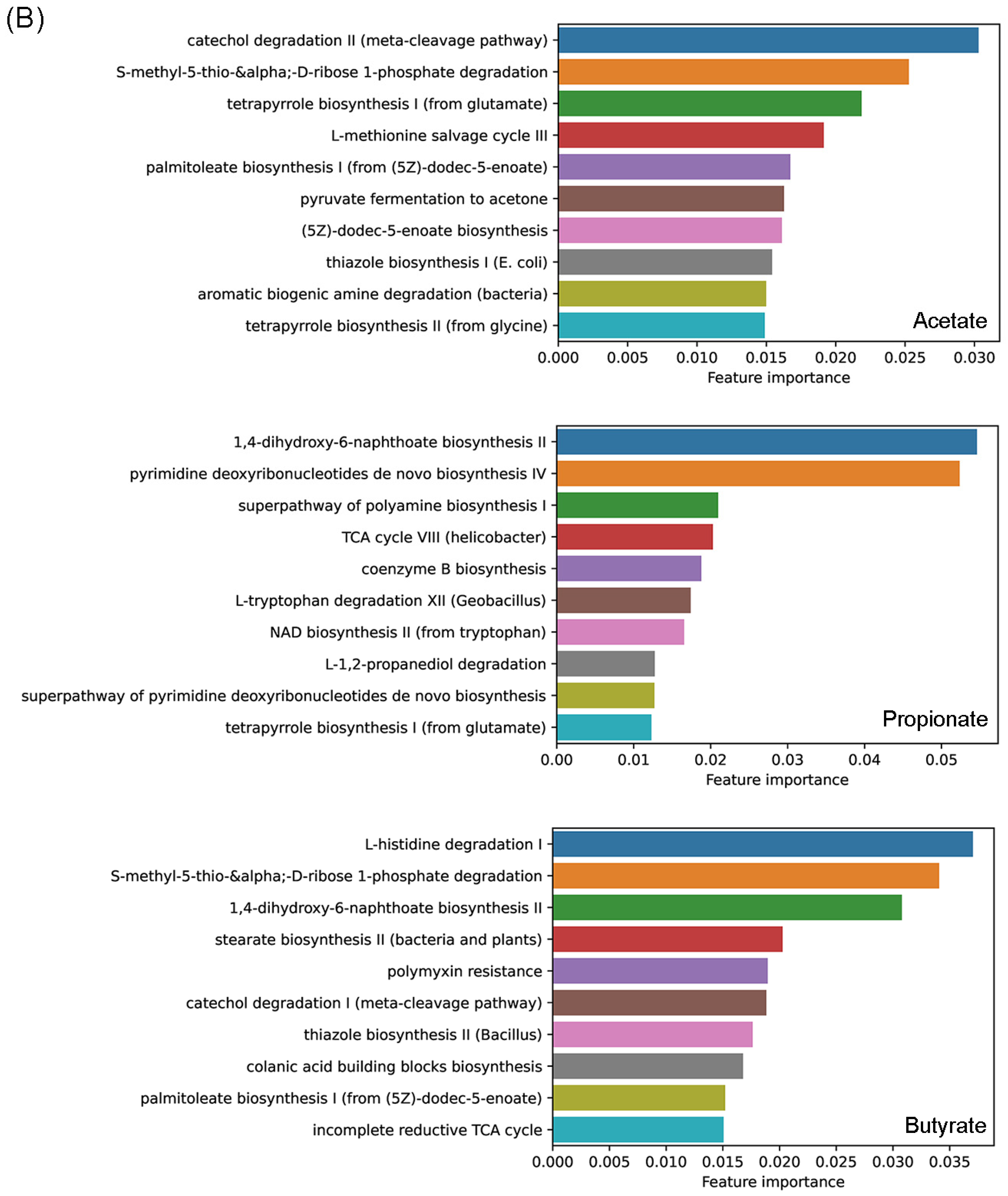
3.4. Effects of Prebiotics Treatments on the Abundance of Genera and Predicted Metabolic Activities
3.5. Predicting Effects of Prebiotics with Machine-Learning Techniques
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gibson, G.R.; Roberfroid, M.B. Dietary Modulation of the Human Colonic Microbiota: Introducing the Concept of Prebiotics. J. Nutr. 1995, 125, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Khangwal, I.; Shukla, P. Prospecting prebiotics, innovative evaluation methods, and their health applications: A review. 3 Biotech 2019, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Davani-Davari, D.; Negahdaripour, M.; Karimzadeh, I.; Seifan, M.; Mohkam, M.; Masoumi, S.J.; Berenjian, A.; Ghasemi, Y. Prebiotics: Definition, Types, Sources, Mechanisms, and Clinical Applications. Foods 2019, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef]
- Canani, R.B.; Costanzo, M.D.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef]
- van der Hee, B.; Wells, J.M. Microbial Regulation of Host Physiology by Short-chain Fatty Acids. Trends Microbiol. 2021, 29, 700–712. [Google Scholar] [CrossRef]
- Nguyen, T.L.A.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis. Model Mech. 2015, 8, 1–16. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Senghor, B.; Sokhna, C.; Ruimy, R.; Lagier, J.-C. Gut microbiota diversity according to dietary habits and geographical provenance. Hum. Microbiome J. 2018, 7–8, 1–9. [Google Scholar] [CrossRef]
- Cryan, J.F.; O′Riordan, K.J.; Sandhu, K.; Peterson, V.; Dinan, T.G. The gut microbiome in neurological disorders. Lancet Neurol. 2020, 19, 179–194. [Google Scholar] [CrossRef]
- De Angelis, M.; Ferrocino, I.; Calabrese, F.M.; De Filippis, F.; Cavallo, N.; Siragusa, S.; Rampelli, S.; Di Cagno, R.; Rantsiou, K.; Vannini, L.; et al. Diet influences the functions of the human intestinal microbiome. Sci. Rep. 2020, 10, 4247. [Google Scholar] [CrossRef] [PubMed]
- Brodkorb, A.; Egger, L.; Alminger, M.; Alvito, P.; Assunção, R.; Ballance, S.; Bohn, T.; Bourlieu-Lacanal, C.; Boutrou, R.; Carrière, F.; et al. INFOGEST static in vitro simulation of gastrointestinal food digestion. Nat. Protoc. 2019, 14, 991–1014. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Abou-Samra, E.; Ning, Z.; Zhang, X.; Mayne, J.; Wang, J.; Cheng, K.; Walker, K.; Stintzi, A.; Figeys, D. An in vitro model maintaining taxon-specific functional activities of the gut microbiome. Nat. Commun. 2019, 10, 4146. [Google Scholar] [CrossRef]
- Singh, V.; Hwang, N.; Ko, G.; Tatsuya, U. Effects of digested Cheonggukjang on human microbiota assessed by in vitro fecal fermentation. J. Microbiol. 2021, 59, 217–227. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Z.; Liu, O. Development and validation of a GC-FID method for quantitative analysis of oleic acid and related fatty acids. J. Pharm. Anal. 2015, 5, 223–230. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2013, 42, D633–D642. [Google Scholar] [CrossRef]
- Westcott, S.L.; Schloss, P.D.; McMahon, K.; Watson, M.; Pollard, K. OptiClust, an Improved Method for Assigning Amplicon-Based Sequence Data to Operational Taxonomic Units. mSphere 2017, 2, e00073-17. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.D.; Macklaim, J.M.; Linn, T.G.; Reid, G.; Gloor, G.B. ANOVA-Like Differential Expression (ALDEx) Analysis for Mixed Population RNA-Seq. PLoS ONE 2013, 8, e67019. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package. R Pakcage Version 2.6-2. 2022. Available online: https://CRAN.R-project.org/package=vegan (accessed on 26 July 2022).
- Zhou, Y.-H.; Gallins, P. A Review and Tutorial of Machine Learning Methods for Microbiome Host Trait Prediction. Front Genet. 2019, 10, 579. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. Xgboost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM Sigkdd International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Hoerl, A.E.; Kennard, R.W. Ridge Regression: Applications to Nonorthogonal Problems. Technometrics 1970, 12, 69–82. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. B 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Altman, N.S. An Introduction to Kernel and Nearest-Neighbor Nonparametric Regression. Am. Stat. 1992, 46, 175–185. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Manabe, N.; Yamaguchi, Y. 3D Structural Insights into β-Glucans and Their Binding Proteins. Int. J. Mol. Sci. 2021, 22, 1578. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Muthuramalingam, K.; Kim, Y.M.; Park, S.; Kim, S.H.; Lee, J.; Hyun, C.; Unno, T.; Cho, M. Synbiotic supplementation with prebiotic Schizophyllum commune derived β-(1,3/1,6)-glucan and probiotic concoction benefits gut microbiota and its associated metabolic activities. Appl. Biol. Chem. 2021, 64, 1–10. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Zhou, L.; Fu, Z.; Zhang, B.; Li, Q.; Pan, L.; Zhou, C.; Zhao, Q.; Shang, Q.; Yu, G. Enterotype-Specific Effect of Human Gut Microbiota on the Fermentation of Marine Algae Oligosaccharides: A Preliminary Proof-of-Concept In Vitro Study. Polymers 2022, 14, 770. [Google Scholar] [CrossRef] [PubMed]
- Chaban, V. Irritable Bowel Syndrome: Novel Concepts for Research and Treatment; IntechOpen: London, UK, 2016. [Google Scholar]
- Lee, I.-S.; Kang, K.S.; Kim, S.-Y. Panax ginseng Pharmacopuncture: Current Status of the Research and Future Challenges. Biomolecules 2020, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Couvigny, B.; de Wouters, T.; Kaci, G.; Jacouton, E.; Delorme, C.; Doré, J.; Renault, P.; Blottière, H.M.; Guédon, E.; Lapaque, N. Commensal Streptococcus salivarius Modulates PPARγ Transcriptional Activity in Human Intestinal Epithelial Cells. PLoS ONE 2015, 10, e0125371. [Google Scholar] [CrossRef]
- Boer, C.G.; Radjabzadeh, D.; Medina-Gomez, C.; Garmaeva, S.; Schiphof, D.; Arp, P.; Koet, T.; Kurilshikov, A.; Fu, J.; Ikram, M.A.; et al. Intestinal microbiome composition and its relation to joint pain and inflammation. Nat. Commun. 2019, 10, 4881. [Google Scholar] [CrossRef]
- Martinović, A.; Cocuzzi, R.; Arioli, S.; Mora, D. Streptococcus thermophilus: To Survive, or Not to Survive the Gastrointestinal Tract, That Is the Question! Nutrients 2020, 12, 2175. [Google Scholar] [CrossRef]
- Præsteng, K.E.; Pope, P.B.; Cann, I.K.O.; Mackie, R.I.; Mathiesen, S.D.; Folkow, L.P.; Eijsink, V.G.H.; Sundset, M.A. Probiotic Dosing of Ruminococcus flavefaciens Affects Rumen Microbiome Structure and Function in Reindeer. Microb. Ecol. 2013, 66, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 2014, 74, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, J.; Fuentes, S.; Grievink, W.; van Niftrik, L.; Tindall, B.J.; Timmerman, H.M.; Rijkers, G.T.; Smidt, H. Characterization of Romboutsia ilealis gen. nov., sp. nov., isolated from the gastro-intestinal tract of a rat, and proposal for the reclassification of five closely related members of the genus Clostridium into the genera Romboutsia gen. nov., Intestinibacter gen. nov., Terrisporobacter gen. nov. and Asaccharospora gen. nov. Int. J. Syst. Evol. Microbiol. 2014, 64, 1600–1616. [Google Scholar] [CrossRef] [PubMed]
- Ricaboni, D.; Mailhe, M.; Khelaifia, S.; Raoult, D.; Million, M. Romboutsia timonensis, a new species isolated from human gut. New Microbes New Infect. 2016, 12, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Bojović, K.; Ignjatović, Đ.; Bajic, S.S.; Milutinović, D.V.; Tomić, M.; Golić, N.; Tolinački, M. Gut Microbiota Dysbiosis Associated With Altered Production of Short Chain Fatty Acids in Children with Neurodevelopmental Disorders. Front. Cell. Infect. Microbiol. 2020, 10, 223. [Google Scholar] [CrossRef]
- Bentinger, M.; Brismar, K.; Dallner, G. The antioxidant role of coenzyme Q. Mitochondrion 2007, 7, S41–S50. [Google Scholar] [CrossRef]
- Lenaz, G.; Fato, R.; Formiggini, G.; Genova, M.L. The role of Coenzyme Q in mitochondrial electron transport. Mitochondrion 2007, 7, S8–S33. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, H.; Jeon, D.; Unno, T. Evaluation of Prebiotics through an In Vitro Gastrointestinal Digestion and Fecal Fermentation Experiment: Further Idea on the Implementation of Machine Learning Technique. Foods 2022, 11, 2490. https://doi.org/10.3390/foods11162490
Song H, Jeon D, Unno T. Evaluation of Prebiotics through an In Vitro Gastrointestinal Digestion and Fecal Fermentation Experiment: Further Idea on the Implementation of Machine Learning Technique. Foods. 2022; 11(16):2490. https://doi.org/10.3390/foods11162490
Chicago/Turabian StyleSong, Hokyung, Dabin Jeon, and Tatsuya Unno. 2022. "Evaluation of Prebiotics through an In Vitro Gastrointestinal Digestion and Fecal Fermentation Experiment: Further Idea on the Implementation of Machine Learning Technique" Foods 11, no. 16: 2490. https://doi.org/10.3390/foods11162490