Deformation Measurements of Neuronal Excitability Using Incoherent Holography Lattice Light-Sheet Microscopy (IHLLS)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
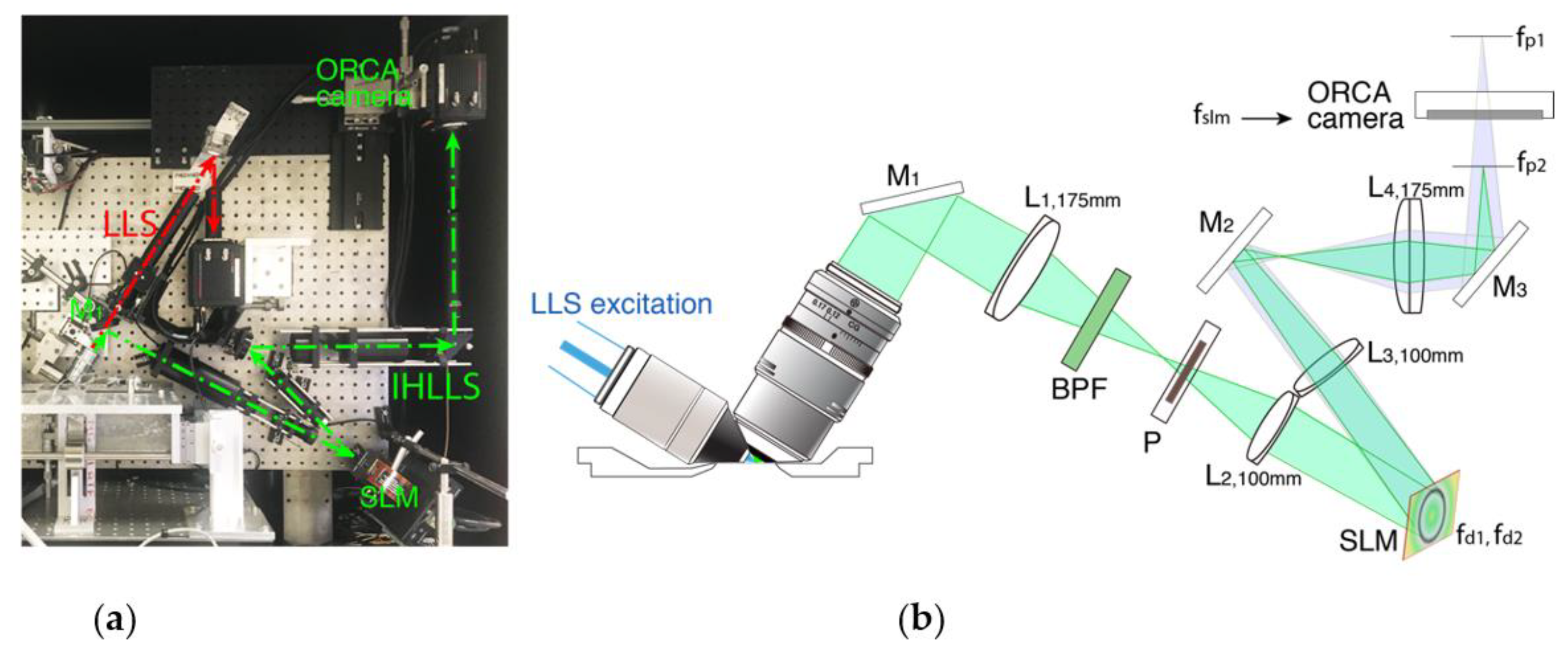
2. Materials and Methods
2.1. System Control
2.2. Sample Preparation
3. Results
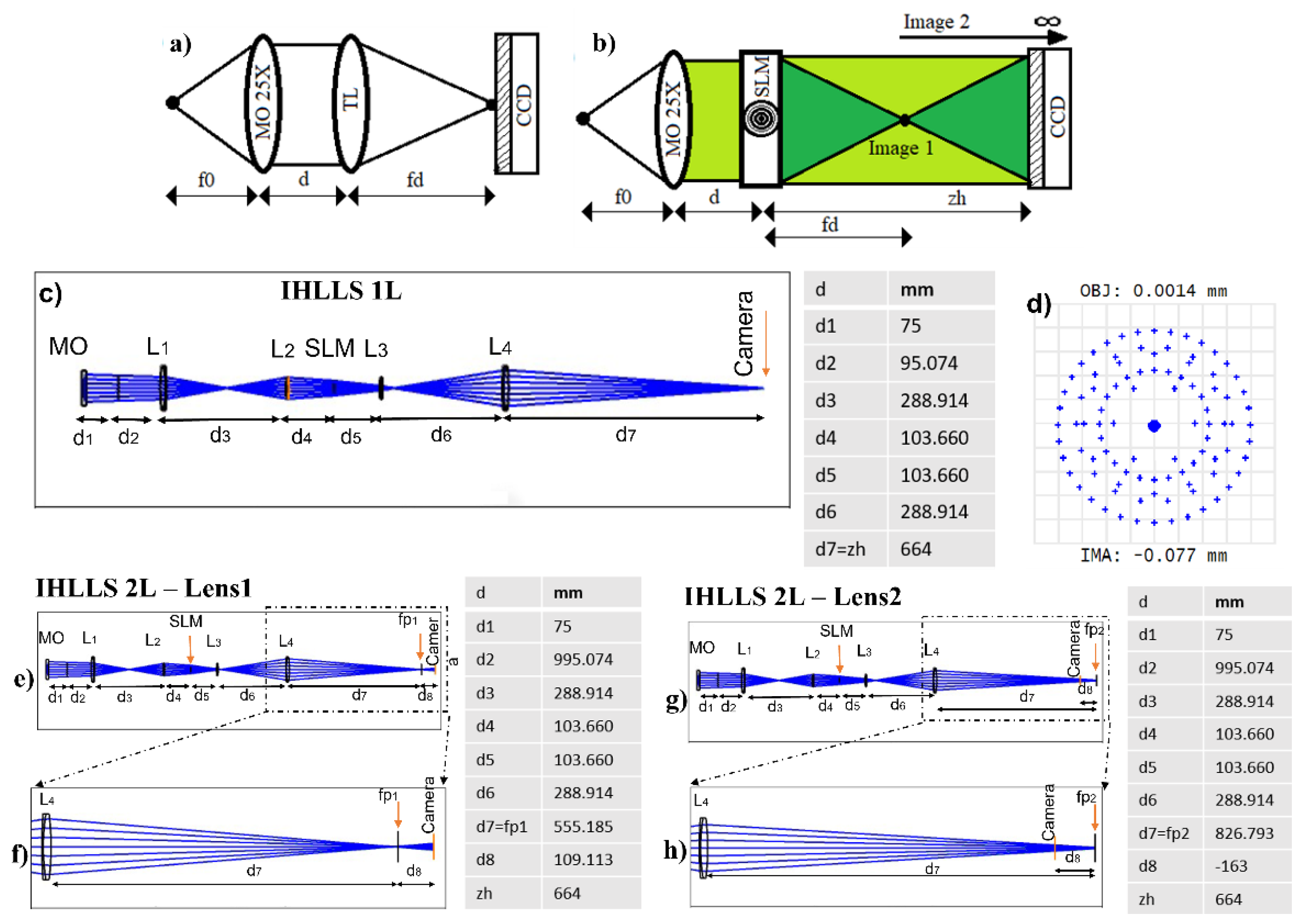
3.1. Fresnel Incoherent Correlation Holography (FINCH) with Dual Lens Adapted for IHLLS
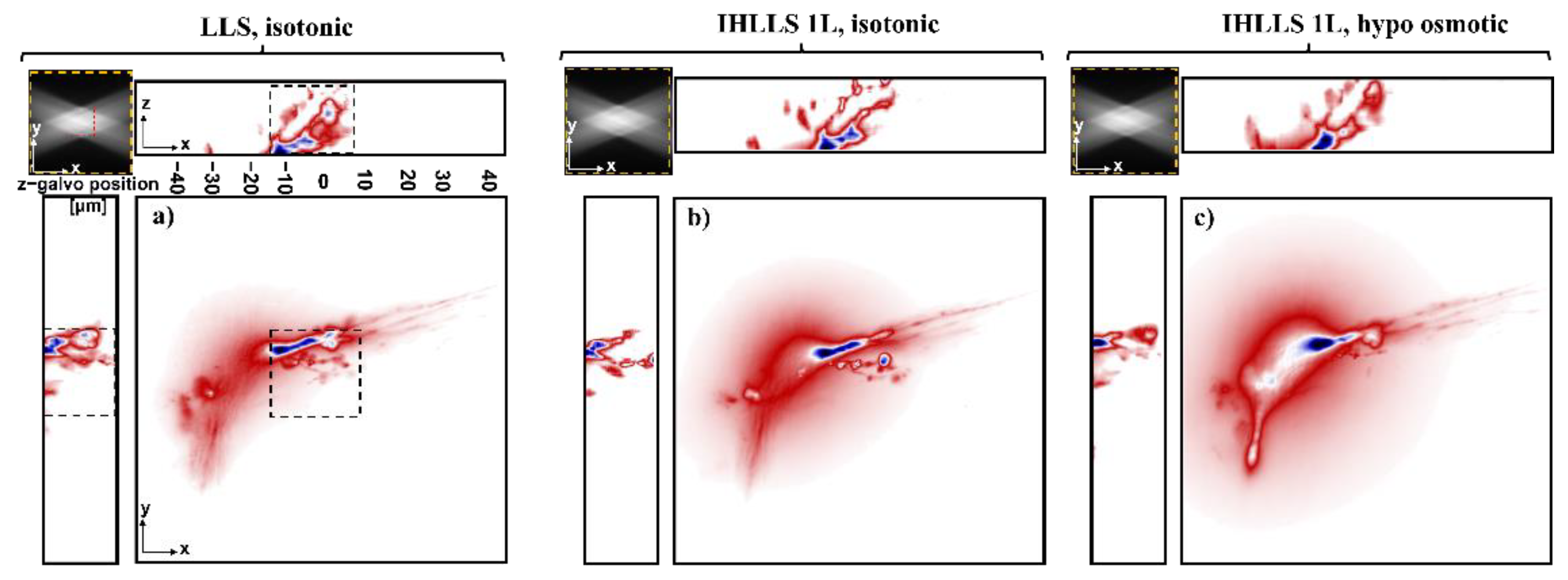
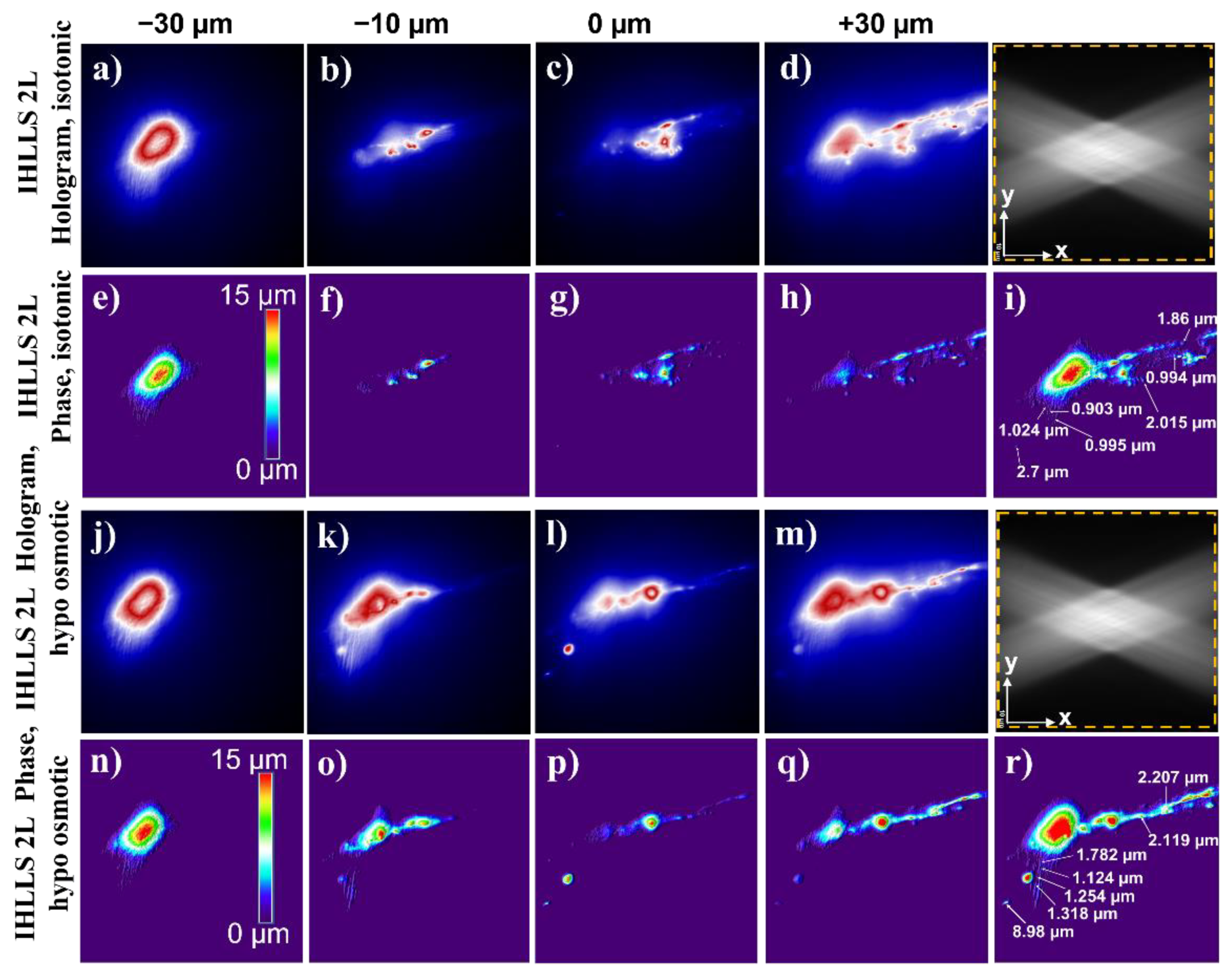
3.2. Imaging Neurons
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Julius, D.; Nathans, J. Signaling by sensory receptors. Cold Spring Harb. Perspect. Biol. 2012, 4, a005991. [Google Scholar] [CrossRef] [Green Version]
- Bader, D.L.; Knight, M.M. Biomechanical analysis of structural deformation in living cells. Med Biol. Eng. Comput. 2008, 46, 951–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lankford, C.K.; Laird, J.G.; Inamdar, S.M.; Baker, S.A. A Comparison of the Primary Sensory Neurons Used in Olfaction and Vision. Front. Cell. Neurosci. 2020, 14, 595523. [Google Scholar] [CrossRef] [PubMed]
- Hodgkin, A.L.; Huxley, A.F. A Quantitative Description of Membrane Current and Its Application to Conduction and Excitation in Nerve. J. Physiol. 1952, 117, 500–544. [Google Scholar] [CrossRef] [PubMed]
- Knöpfel, T.; Tomita, K.; Shimazaki, R.; Sakai, R. Optical recordings of membrane potential using genetically targeted voltage-sensitive fluorescent proteins. Methods 2003, 30, 42–48. [Google Scholar] [CrossRef]
- Fillafer, C.; Mussel, M.; Muchowski, J.; Schneider, M.F. Cell Surface Deformation during an Action Potential. Biophys. J. 2018, 114, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Mueller, J.K.; Tyler, W.J. A quantitative overview of biophysical forces impinging on neural function. Phys. Biol. 2014, 11, 051001. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.-C.; Keleshian, A.M.; Sachs, F. Voltage-induced membrane movement. Nature 2001, 413, 428–432. [Google Scholar] [CrossRef]
- Lindau, M.; Neher, E. Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflügers Arch. 1988, 411, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, J.; Wohland, T. Fluorescence strategies for mapping cell membrane dynamics and structures. APL Bioeng. 2020, 4, 020901. [Google Scholar] [CrossRef]
- Boulanger, J.; Gueudry, C.; Münch, D.; Cinquin, B.; Paul-Gilloteaux, P.; Bardin, S.; Guérin, C.; Senger, F.; Blanchoin, L.; Salamero, J. Fast high-resolution 3D total internal reflection fluorescence microscopy by incidence angle scanning and azimuthal averaging. Proc. Natl. Acad. Sci. USA 2014, 111, 17164–17169. [Google Scholar] [CrossRef] [Green Version]
- Tolde, O.; Gandalovičová, A.; Křížová, A.; Veselý, P.; Chmelík, R.; Rosel, D.; Brábek, J. Quantitative phase imaging unravels new insight into dynamics of mesenchymal and amoeboid cancer cell invasion. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Anvari, B. Characterization of the Viscoelastic Properties of Ovarian Cancer Cells Membranes by Optical Tweezers and Quantitative Phase Imaging. Front. Phys. 2020, 8. [Google Scholar] [CrossRef]
- Jaferzadeh, K.; Sim, M.; Kim, N.; Moon, I. Quantitative analysis of three-dimensional morphology and membrane dynamics of red blood cells during temperature elevation. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaferzadeh, K.; Moon, I.; Bardyn, M.; Prudent, M.; Tissot, J.-D.; Rappaz, B.; Javidi, B.; Turcatti, G.; Marquet, P. Quantification of stored red blood cell fluctuations by time-lapse holographic cell imaging. Biomed. Opt. Express 2018, 9, 4714–4729. [Google Scholar] [CrossRef]
- Rappaz, B.; Barbul, A.; Hoffmann, A.; Boss, D.; Korenstein, R.; Depeursinge, C.; Magistretti, P.J.; Marquet, P. Spatial analysis of erythrocyte membrane fluctuations by digital holographic microscopy. Blood Cells Mol. Dis. 2009, 42, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Mohanty, S.; Zhang, J.; Genc, S.; Kim, M.K.; Berns, M.; Chen, Z. Digital holographic microscopy for quantitative cell dynamic evaluation during laser microsurgery. Opt. Express 2009, 17, 12031–12038. [Google Scholar] [CrossRef] [Green Version]
- Gabor, D. A New Microscopic Principle. Nature 1948, 161, 777–778. [Google Scholar] [CrossRef] [PubMed]
- Mann, C.J.; Yu, L.; Lo, C.-M.; Kim, M.K. High-resolution quantitative phase-contrast microscopy by digital holography. Opt. Express 2005, 13, 8693–8698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khmaladze, A.; Kim, M.; Lo, C.M. Phase imaging of cells by simultaneous dual-wavelength reflection digital holography. Opt. Express 2008, 16, 10900–10911. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K. Principles and techniques of digital holographic microscopy. J. Photon. Energy 2010, 1, 018005. [Google Scholar] [CrossRef] [Green Version]
- Ferraro, P. Coherent Light Microscopy: Imaging and Quantitative Phase Analysis; Ferraro, P., Wax, A., Zalevsky, Z., Eds.; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Rosen, J.; Kelner, R. Modified Lagrange invariants and their role in determining transverse and axial imaging resolutions of self-interference incoherent holographic systems. Opt. Express 2014, 22, 29048–29066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potcoava, M.; Mann, C.; Art, J.; Alford, S. Spatio-temporal performance enhancement in an incoherent holography lattice light-sheet microscope (IHLLS). Opt. Express 2021, 29, 23888–23901. [Google Scholar] [CrossRef]
- Gao, L.; Shao, L.; Chen, B.-C.; Betzig, E. 3D live fluorescence imaging of cellular dynamics using Bessel beam plane illumination microscopy. Nat. Protoc. 2014, 9, 1083–1101. [Google Scholar] [CrossRef]
- Chen, B.C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A.; Liu, Z.; et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef] [Green Version]
- Rosen, J.; Brooker, G. Digital spatially incoherent Fresnel holography. Opt. Lett. 2007, 32, 912–914. [Google Scholar] [CrossRef]
- Rosen, J.; Brooker, G. Non-scanning motionless fluorescence three-dimensional holographic microscopy. Nat. Photon. 2008, 2, 190–195. [Google Scholar] [CrossRef]
- Katz, B.; Rosen, J.; Kelner, R.; Brooker, G. Enhanced resolution and throughput of Fresnel incoherent correlation holography (FINCH) using dual diffractive lenses on a spatial light modulator (SLM). Opt. Express 2012, 20, 9109–9121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooker, G.; Siegel, N.; Wang, V.; Rosen, J. Optimal resolution in Fresnel incoherent correlation holographic fluorescence microscopy. Opt. Express 2011, 19, 5047–5062. [Google Scholar] [CrossRef] [Green Version]
- Bouchal, P.; Bouchal, Z. Wide-field common-path incoherent correlation microscopy with a perfect overlapping of interfering beams. J. Eur. Opt. Soc. Rapid Publ. 2013, 8, 13011. [Google Scholar] [CrossRef] [Green Version]
- Rosen, J.; Siegel, N.; Brooker, G. Theoretical and experimental demonstration of resolution beyond the Rayleigh limit by FINCH fluorescence microscopic imaging. Opt. Express 2011, 19, 26249–26268. [Google Scholar] [CrossRef] [PubMed]
- Rosen, J.; Brooken, G. Fresnel incoherent correlation holography (FINCH): A review of research. Adv. Opt. Technol. 2012, 1, 151–169. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potcoava, M.; Art, J.; Alford, S.; Mann, C. Deformation Measurements of Neuronal Excitability Using Incoherent Holography Lattice Light-Sheet Microscopy (IHLLS). Photonics 2021, 8, 383. https://doi.org/10.3390/photonics8090383
Potcoava M, Art J, Alford S, Mann C. Deformation Measurements of Neuronal Excitability Using Incoherent Holography Lattice Light-Sheet Microscopy (IHLLS). Photonics. 2021; 8(9):383. https://doi.org/10.3390/photonics8090383
Chicago/Turabian StylePotcoava, Mariana, Jonathan Art, Simon Alford, and Christopher Mann. 2021. "Deformation Measurements of Neuronal Excitability Using Incoherent Holography Lattice Light-Sheet Microscopy (IHLLS)" Photonics 8, no. 9: 383. https://doi.org/10.3390/photonics8090383