
Advanced Process Analytical Technology in Combination with Process Modeling for Endpoint and Model Parameter Determination in Lyophilization Process Design and Optimization



Abstract
:1. Introduction
- -
- High storage stability;
- -
- Gentle removal of water;
- -
- Short reconstitution time;
- -
- Accurate dosing;
- -
- Aseptic handling.
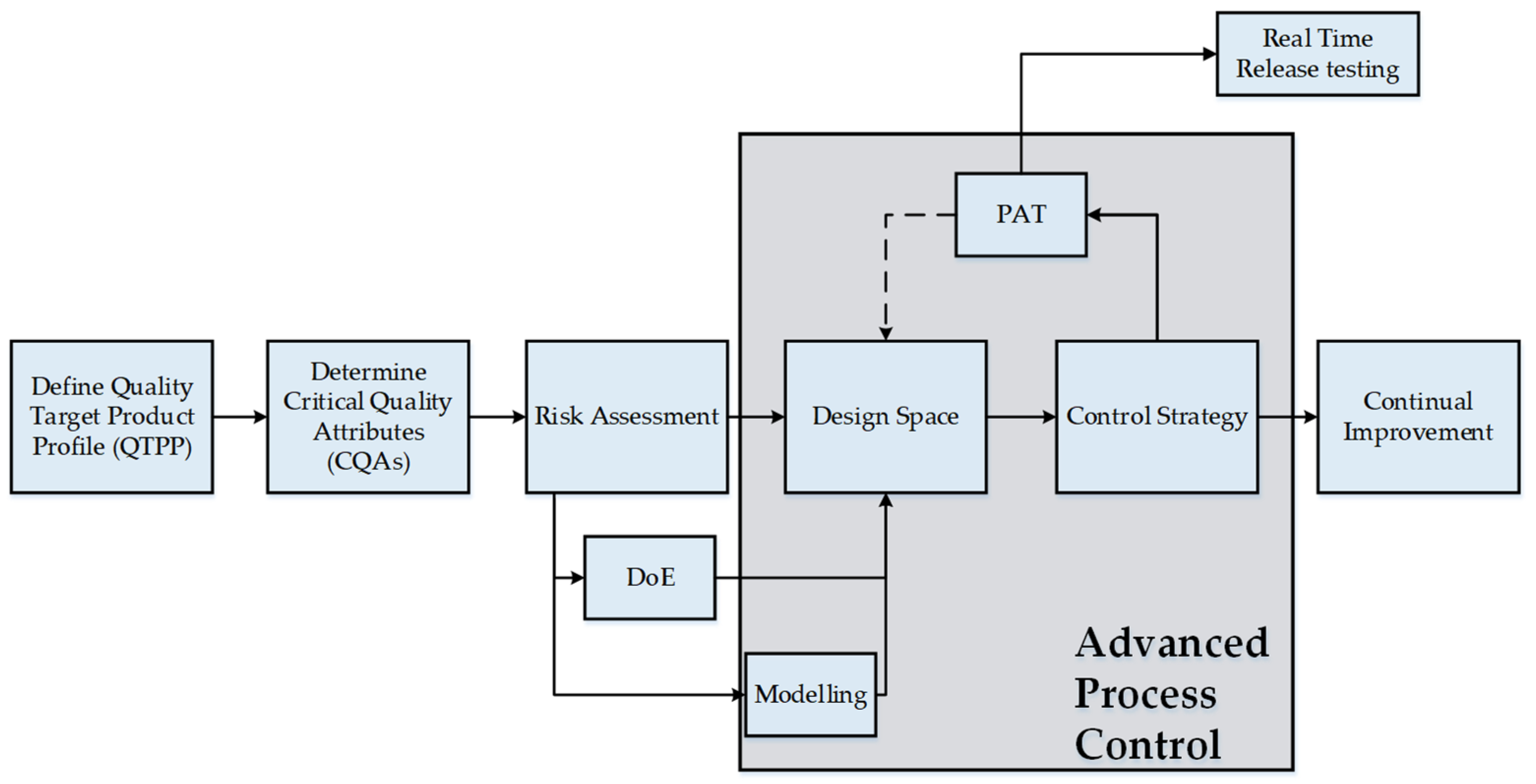
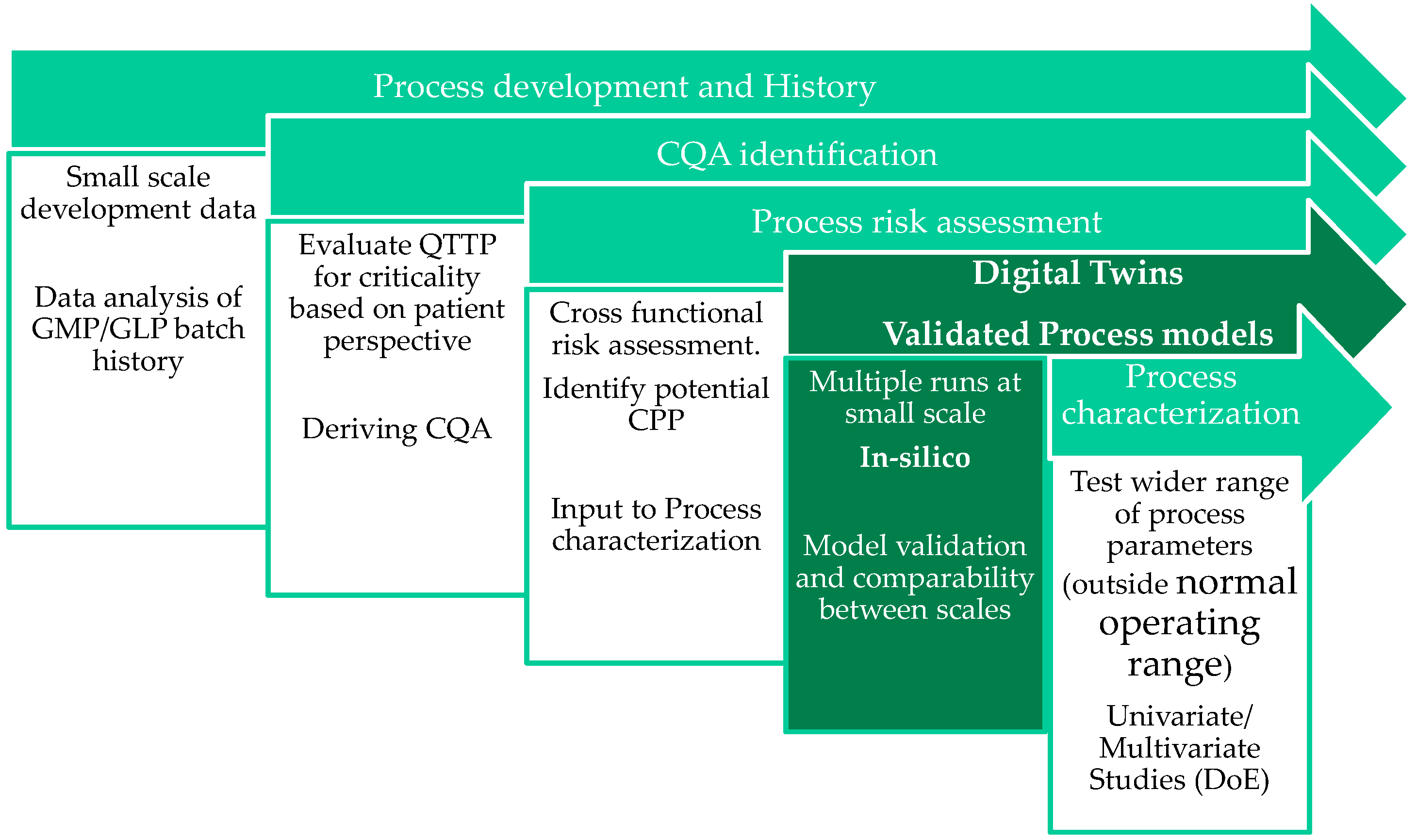
2. Quality by Design in Lyophilization
- -
- Product integrity and stability;
- -
- Drug potency;
- -
- Reconstitution time;
- -
- Cake appearance.
- -
- Freezing: temperature, rate;
- -
- Annealing: temperature, time;
- -
- Primary drying: temperature, pressure, duration;
- -
- Secondary drying: temperature, pressure, duration.
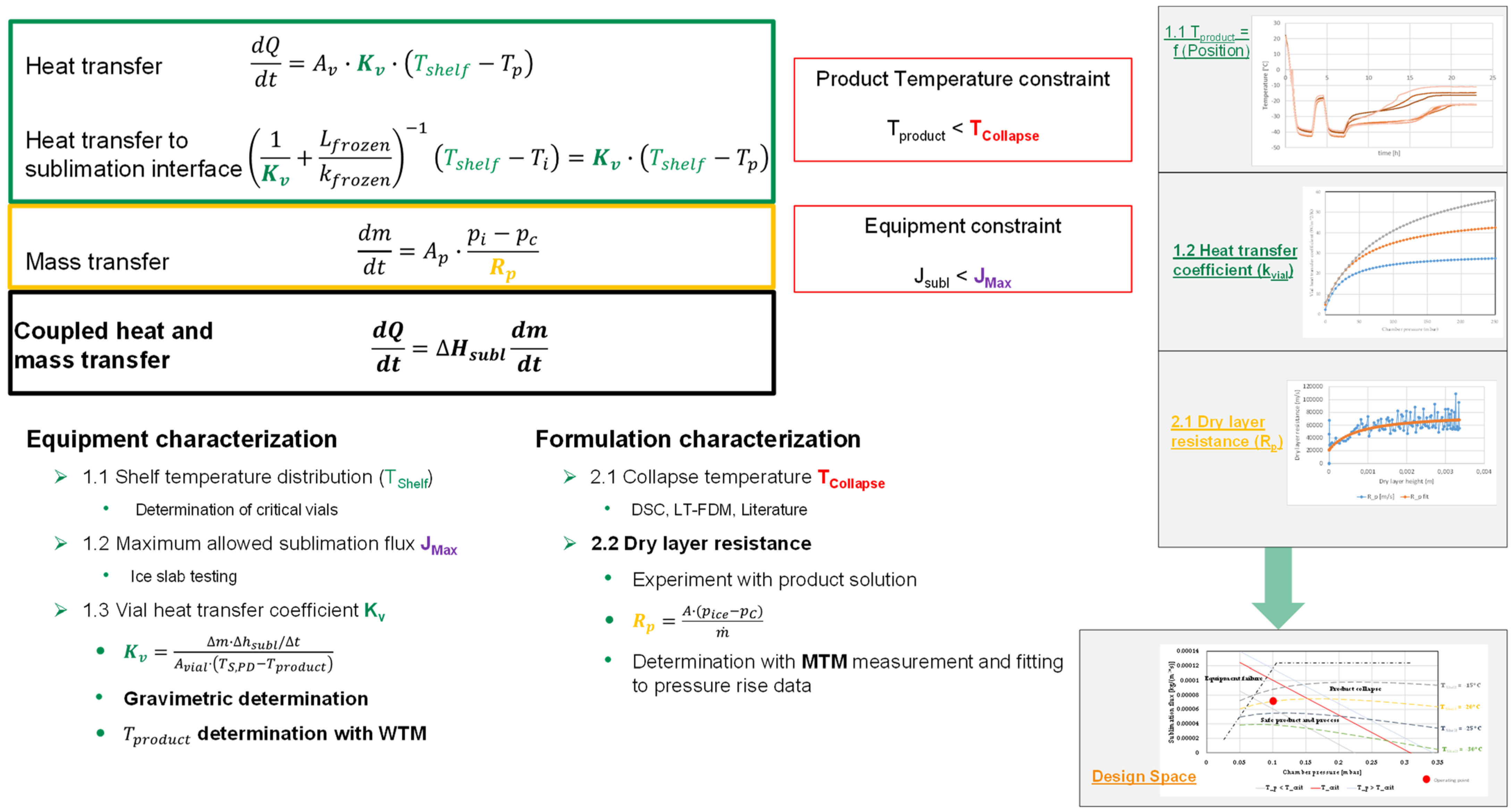
3. Steady State Modeling of Primary Drying Phase
- -
- Allows process design;
- -
- Deepens process understanding;
- -
- Failure analysis;
- -
- Accelerated process development;
- -
- Technology transfer and Scale up.
- -
- -
- -
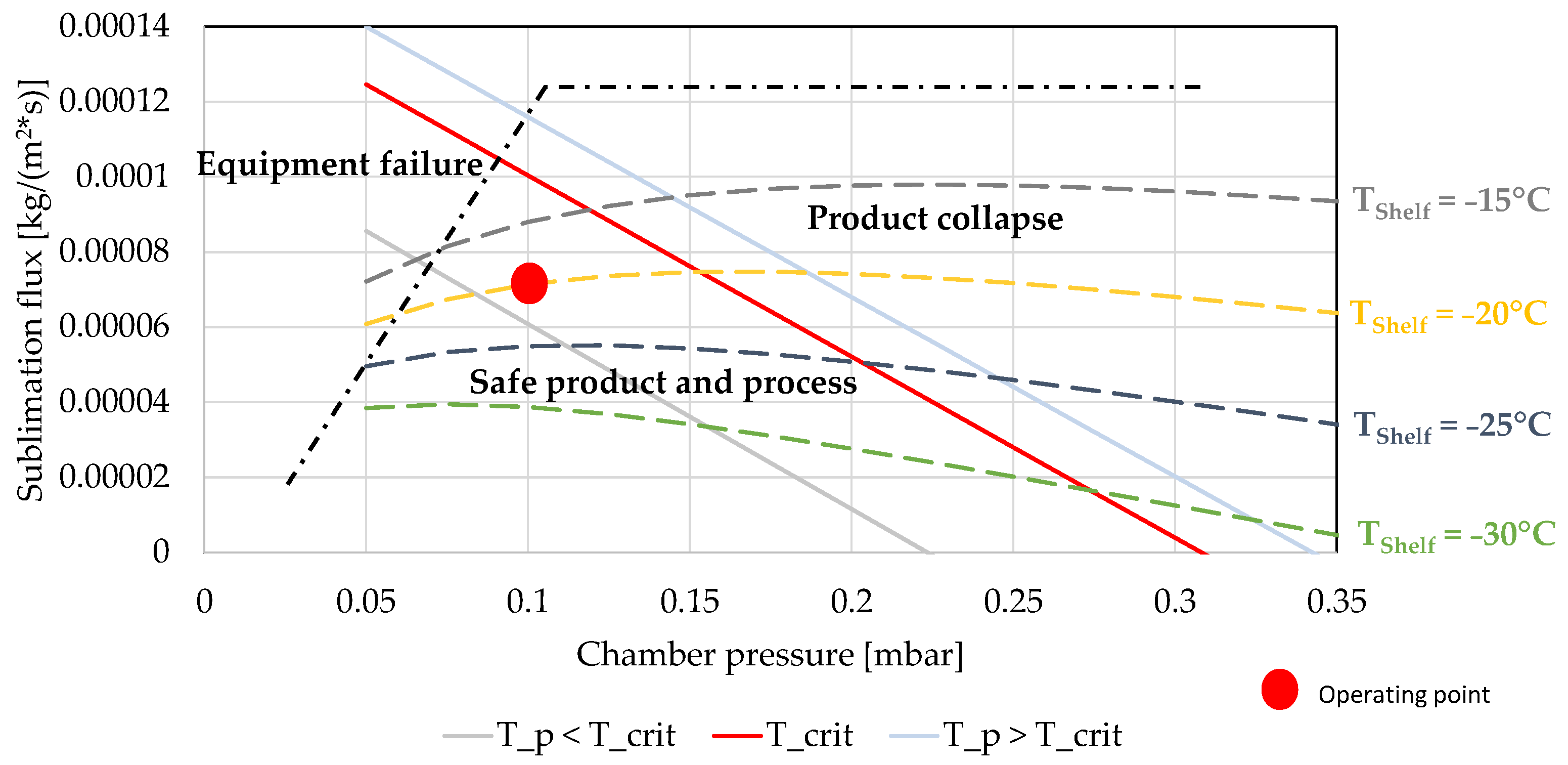
4. Construction of the Design Space
5. Process Analytical Technology for Lyophilization
- -
- TDLAS;
- -
- Mass spectrometry (MS);
- -
- Near-infrared spectroscopy (NIR);
- -
- Raman spectroscopy;
- -
- MTM.
6. Materials and Methods
6.1. Product Mixture and Instruments
6.2. Freeze-Drying Equipment
6.3. Experimental Runs
6.4. Software
6.5. Overall Vial Heat Transfer Coefficient
6.6. Dry Layer Resistance
7. Results
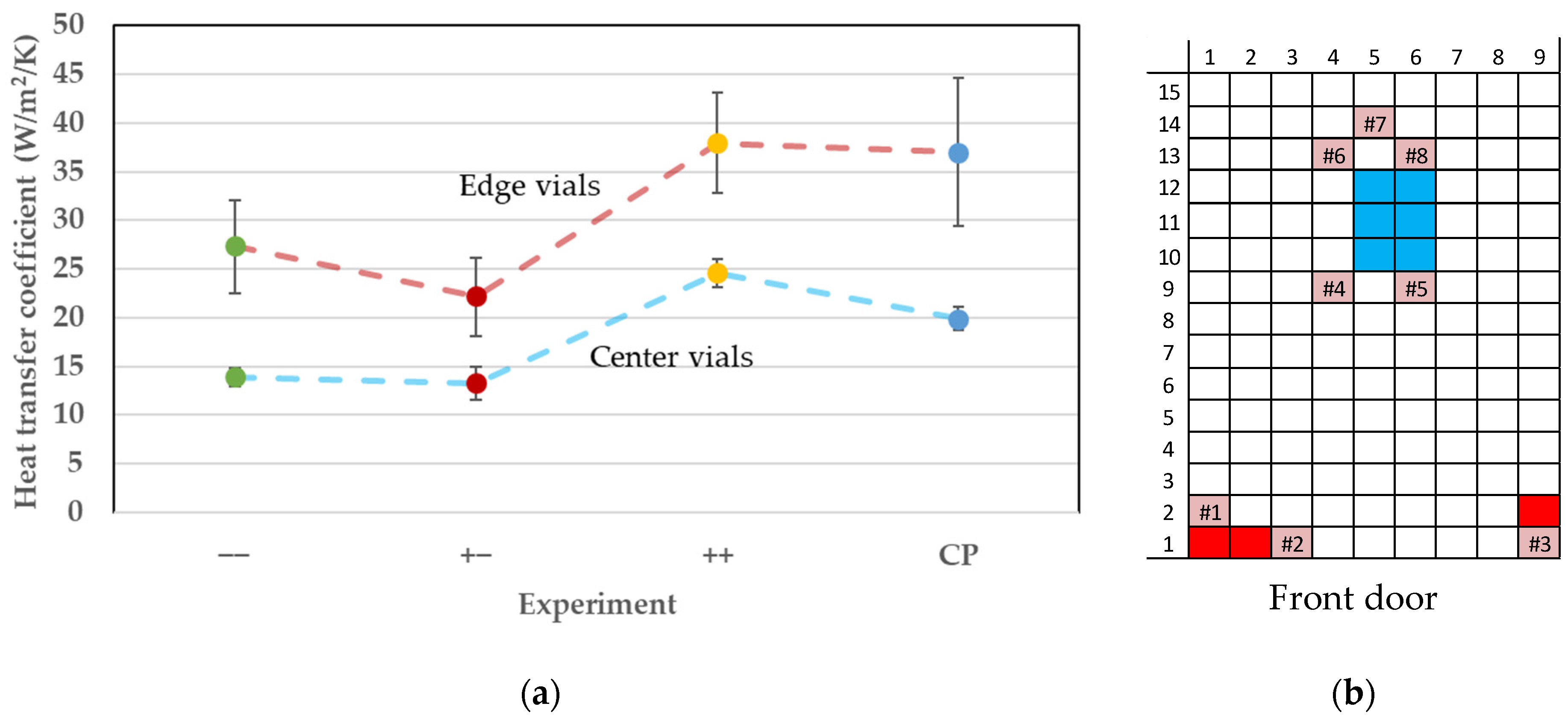
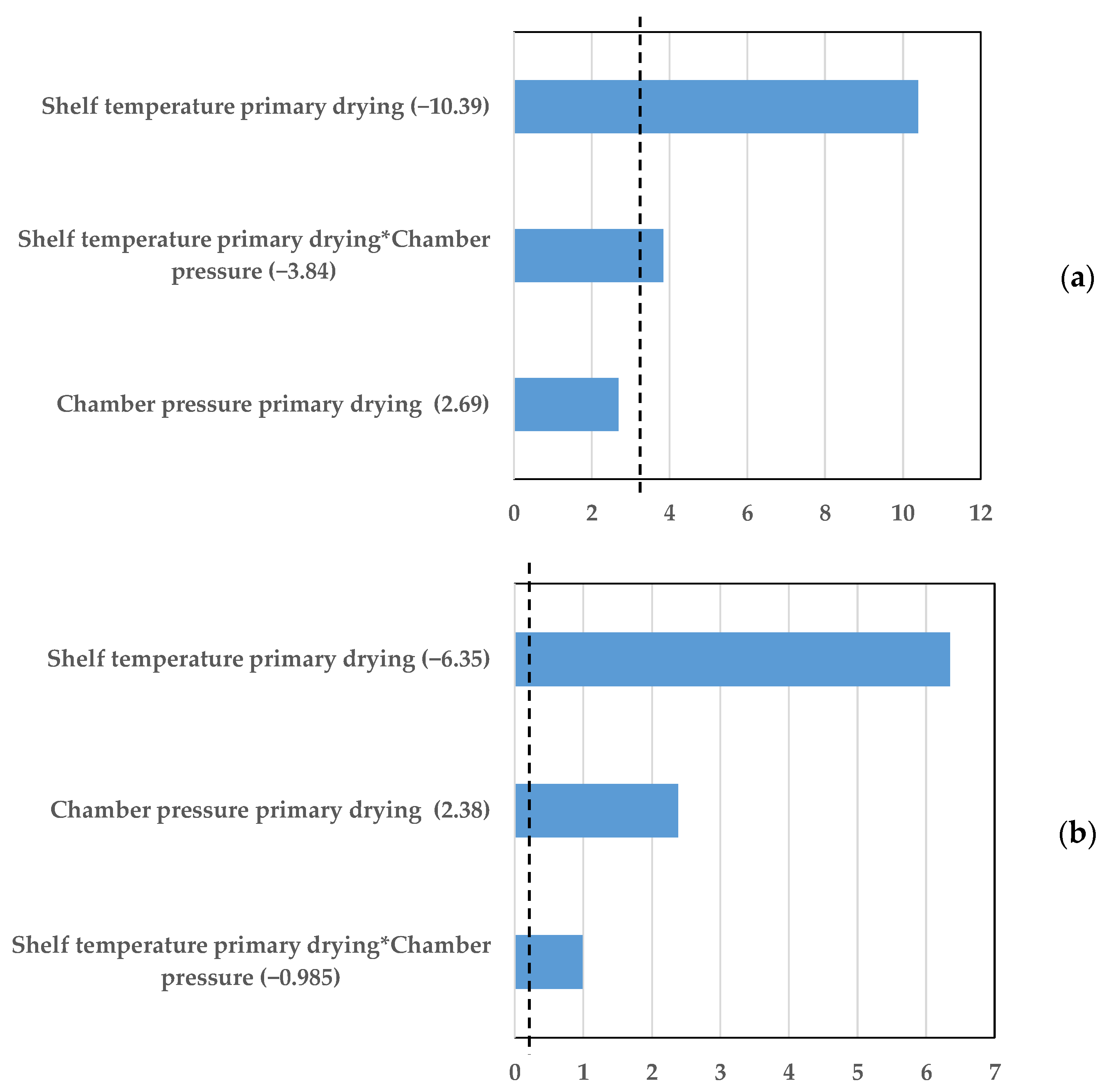
7.1. Vial Heat Transfer Coefficient
7.2. Dry Layer Resistance
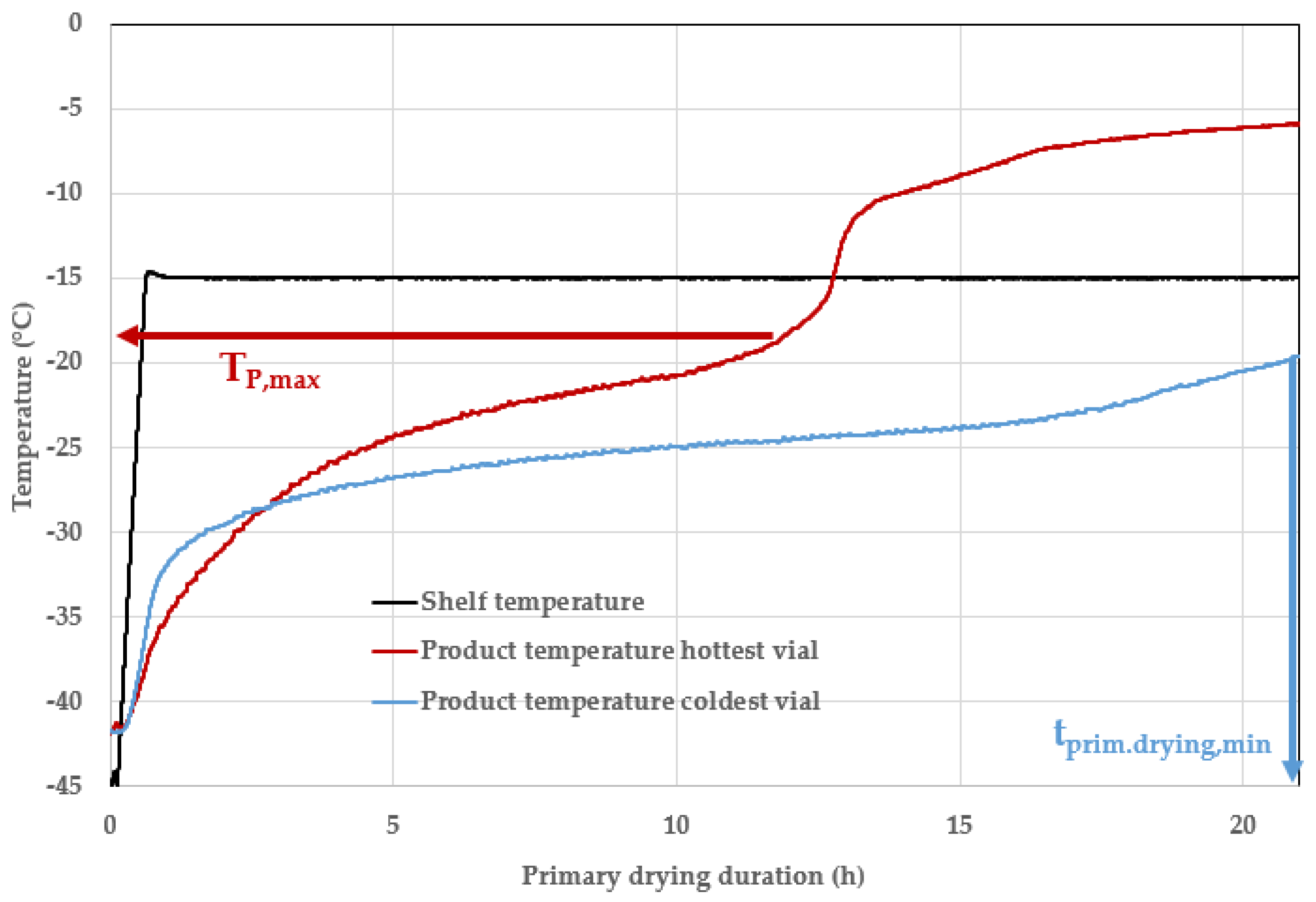
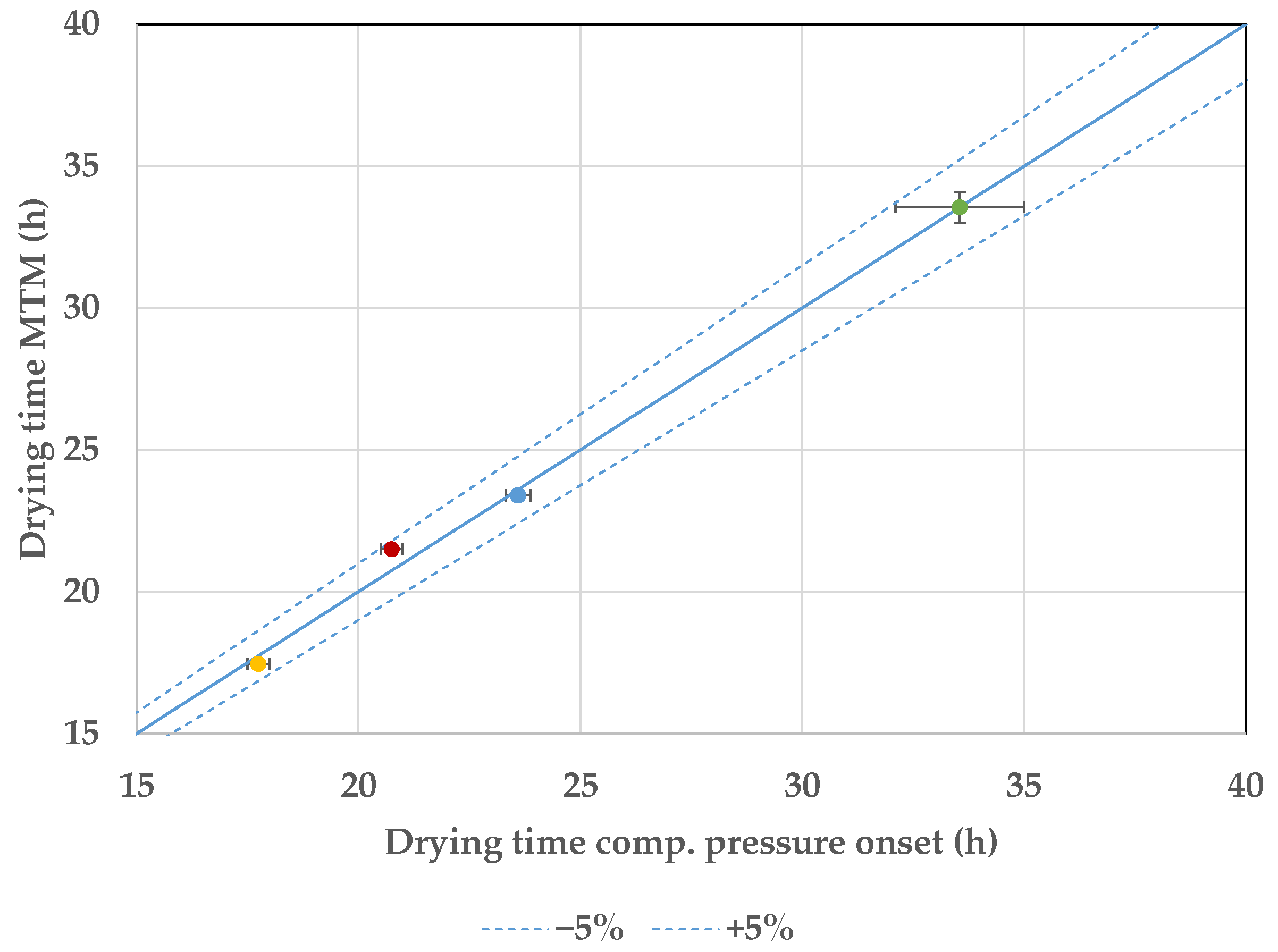
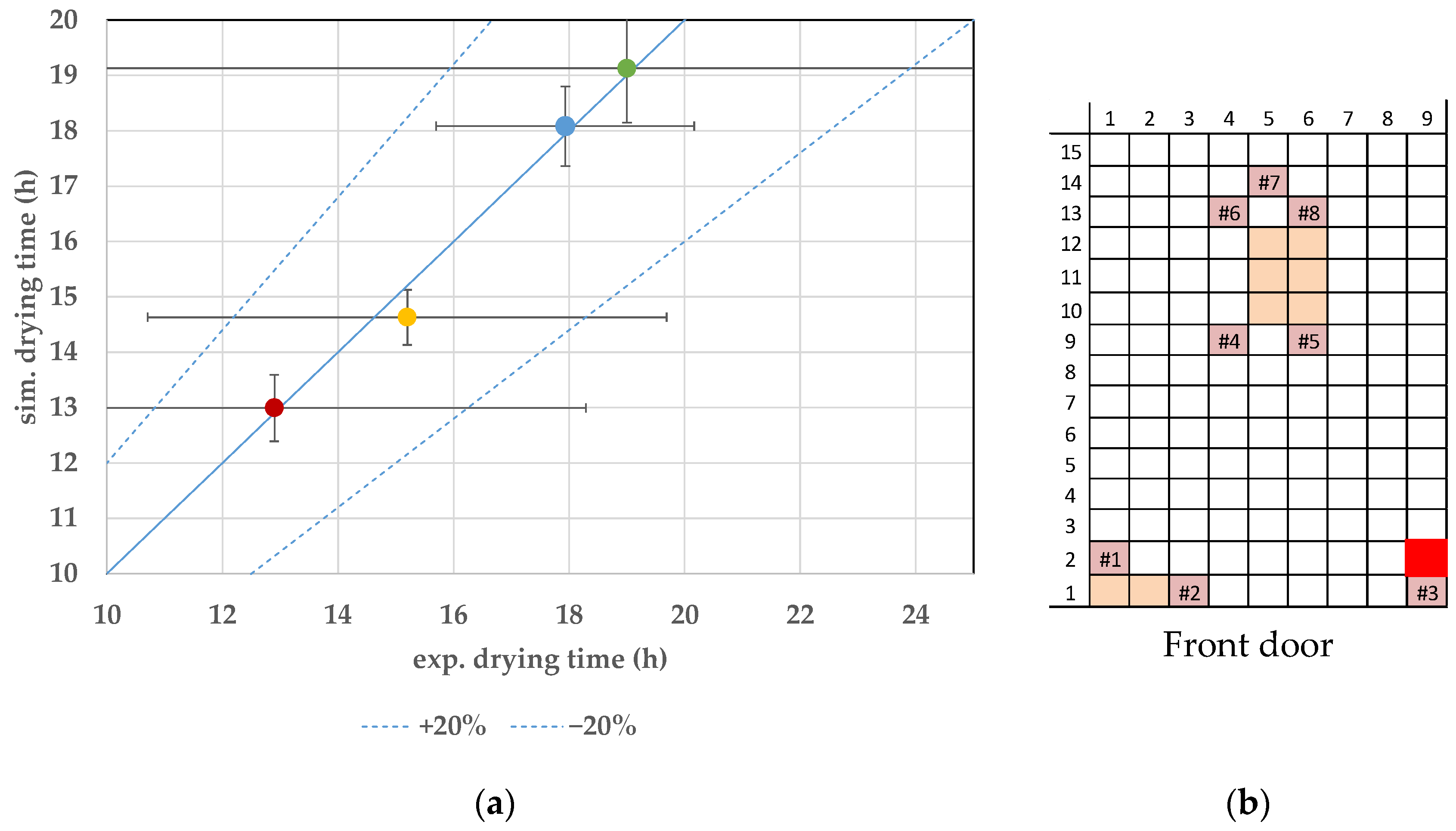
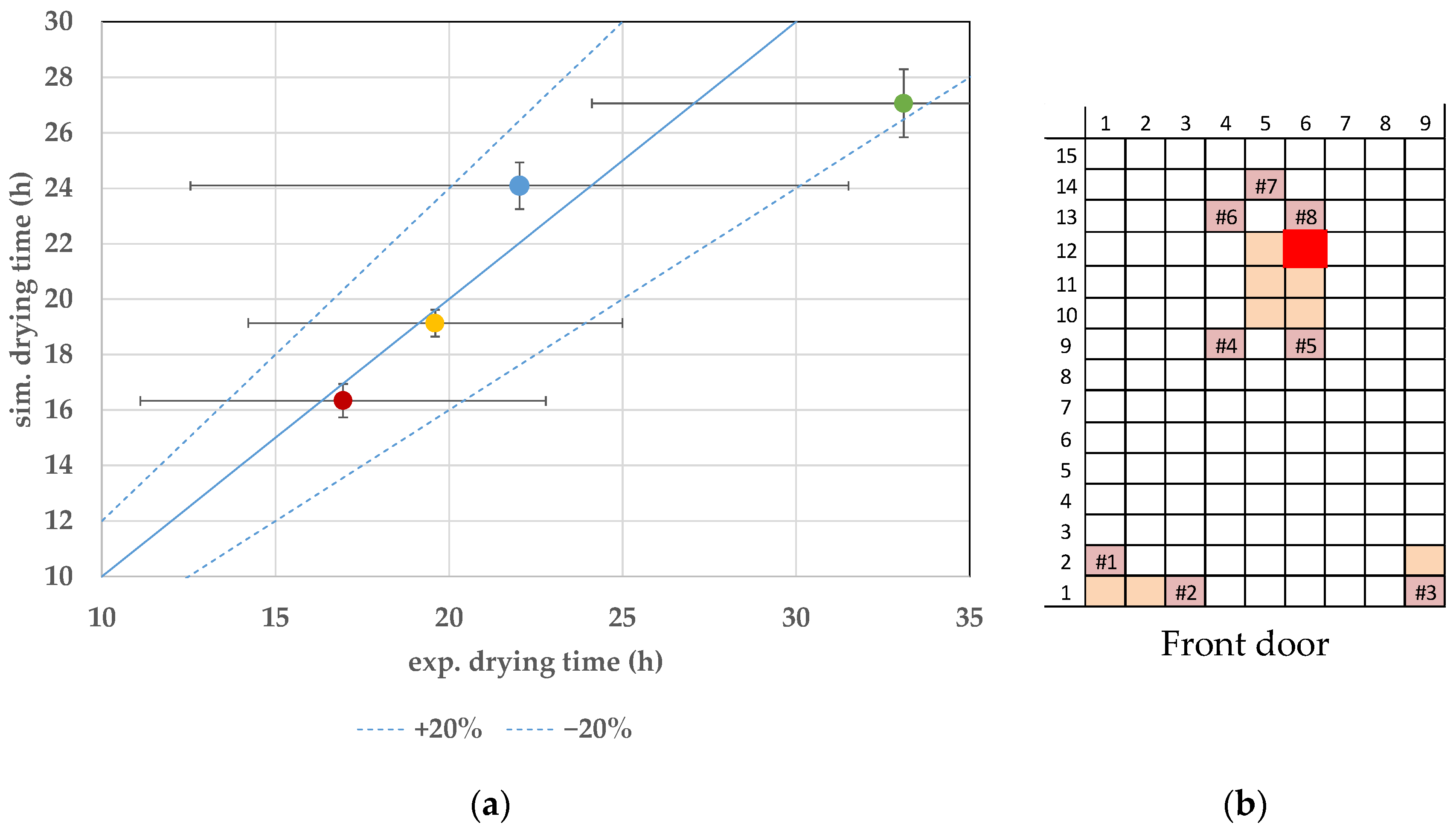
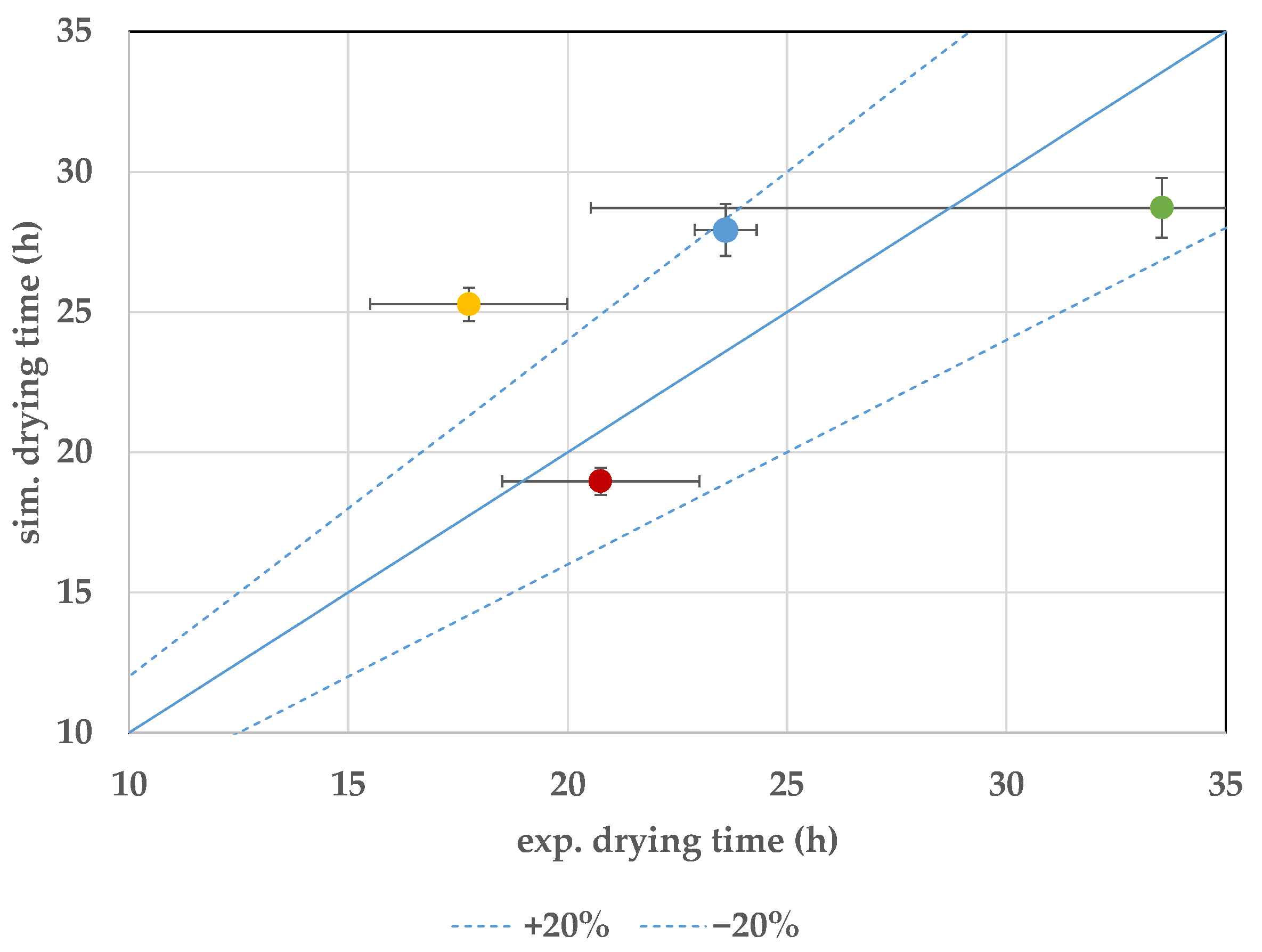
7.3. Endpoint Determination
8. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Symbols and Abbreviations
| Latin symbols | ||
| Ap | Inner-cross sectional area | m2 |
| Av | Outer-cross sectional area | m2 |
| Hsubl | Sublimationenthalpy | J/kg |
| Kv | Vial heat transfer coefficient | J/(m2·K·s) |
| L | Length | m |
| N | Number of vials | - |
| p | Pressure | Pa |
| Q | Heat | J |
| Rp | Dry layer resistance | m/s |
| T | Temperature | K |
| t | Time | s |
| X | Variable for linear pressure increase | Pa/s |
| Greek symbols | ||
| Δ | Difference | - |
| λ | Heat conductivity | W/m/K |
| Indices | ||
| 0 | Value at measurement start | |
| c | Chamber | |
| dry | Dry layer | |
| frozen | Frozen layer | |
| i | Sublimation interface | |
| p | Product | |
| s | Shelf | |
| Abbrevations | ||
| CPP | Critical Process Parameter | |
| CQA | Critical Quality Attributes | |
| DoE | Design of Experiments | |
| FDA | US Food and Drug Administration | |
| FMEA | Failure Mode Effect Analysis | |
| MS | Mass spectrometry | |
| MTM | Manometric temperature measurement | |
| NIR | Near-infrared spectroscopy | |
| PAT | Process Analytical Technology | |
| QbD | Quality by Design | |
| TDLAS | Tunable Diode Laser Adsorption Spectroscopy | |
| WTMplus | Wireless Temperature Measurment plus | |
Appendix A


References
- Butreddy, A.; Dudhipala, N.; Janga, K.Y.; Gaddam, R.P. Lyophilization of Small-Molecule Injectables: An Industry Perspective on Formulation Development, Process Optimization, Scale-Up Challenges, and Drug Product Quality Attributes. AAPS PharmSciTech 2020, 21, 252. [Google Scholar] [CrossRef]
- Depreter, F.; Pilcer, G.; Amighi, K. Inhaled proteins: Challenges and perspectives. Int. J. Pharm. 2013, 447, 251–280. [Google Scholar] [CrossRef]
- Langford, A.; Bhatnagar, B.; Walters, R.; Tchessalov, S.; Ohtake, S. Drying technologies for biopharmaceutical applications: Recent developments and future direction. Dry. Technol. 2018, 36, 677–684. [Google Scholar] [CrossRef]
- Pisano, R.; Arsiccio, A.; Capozzi, L.C.; Trout, B.L. Achieving continuous manufacturing in lyophilization: Technologies and approaches. Eur. J. Pharm. Biopharm. 2019, 142, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Emami, F.; Vatanara, A.; Park, E.J.; Na, D.H. Drying Technologies for the Stability and Bioavailability of Biopharmaceuticals. Pharmaceutics 2018, 10, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA. Lyophilization of Parenteral (7/93). FDA [Online]. 3 November 2018. Available online: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-guides/lyophilization-parenteral-793 (accessed on 3 December 2020).
- Difranco, N. Lyophilization of Pharmaceuticals: An Overview. Lubrizol CDMO [Online]. 8 October 2019. Available online: https://lubrizolcdmo.com/blog/lyophilization-of-pharmaceuticals-an-overview/ (accessed on 3 December 2020).
- Mirasol, F. Lyophilization Presents Complex Challenges. BioPharm Int. 2020, 33, 22–24. [Google Scholar]
- Thomas, F. Changing Perceptions: An Understanding of Lyophilization Advancements. Pharm. Technol. 2019, 43, 32–34. [Google Scholar]
- Kasper, J.C.; Winter, G.; Friess, W. Recent advances and further challenges in lyophilization. Eur. J. Pharm. Biopharm. 2013, 85, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Shimanouchi, T.; Kimura, Y. Recent Development of Optimization of Lyophilization Process. J. Chem. 2019, 2019, 9502856. [Google Scholar] [CrossRef]
- FDA. PAT—A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance. FDA [Online]. 11 June 2020. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/pat-framework-innovative-pharmaceutical-development-manufacturing-and-quality-assurance (accessed on 15 February 2021).
- ICH. Pharamceutical Development Q8 (R2). Available online: https://database.ich.org/sites/default/files/Q8%28R2%29%20Guideline.pdf (accessed on 12 July 2021).
- Helgers, H.; Schmidt, A.; Lohmann, L.J.; Vetter, F.L.; Juckers, A.; Jensch, C.; Mouellef, M.; Zobel-Roos, S.; Strube, J. Towards Autonomous Operation by Advanced Process Control—Process Analytical Technology for Continuous Biologics Antibody Manufacturing. Processes 2021, 9, 172. [Google Scholar] [CrossRef]
- Klepzig, L.S.; Juckers, A.; Knerr, P.; Harms, F.; Strube, J. Digital Twin for Lyophilization by Process Modeling in Manufacturing of Biologics. Processes 2020, 8, 1325. [Google Scholar] [CrossRef]
- Aydin, E.S.; Yucel, O.; Sadikoglu, H. Modelling and simulation of a moving interface problem: Freeze drying of black tea extract. Heat Mass Transf. 2017, 53, 2143–2154. [Google Scholar] [CrossRef]
- Song, C.S.; Nam, J.H. A numerical study on freeze drying characteristics of cylindrical products with and without container. Int. J. Transp. Phenom. 2005, 7, 241–254. [Google Scholar]
- Fissore, D.; Pisano, R.; Barresi, A.A. Applying quality-by-design to develop a coffee freeze-drying process. J. Food Eng. 2014, 123, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Mascarenhas, W.J.; Akay, H.U.; Pikal, M.J. A computational model for finite element analysis of the freeze-drying process. Comput. Methods Appl. Mech. Eng. 1997, 148, 105–124. [Google Scholar] [CrossRef]
- Pikal, M.J.; Mascarenhas, W.J.; Akay, H.U.; Cardon, S.; Bhugra, C.; Jameel, F.; Rambhatla, S. The Nonsteady State Modeling of Freeze Drying: In-Process Product Temperature and Moisture Content Mapping and Pharmaceutical Product Quality Applications. Pharm. Dev. Tech. 2005, 10, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Ravnik, J.; Golobič, I.; Sitar, A.; Avanzo, M.; Irman, Š.; Kočevar, K.; Cegnar, M.; Zadravec, M.; Ramšak, M.; Hriberšek, M. Lyophilization model of mannitol water solution in a laboratory scale lyophilizer. J. Drug Deliv. Sci. Technol. 2018, 45, 28–38. [Google Scholar] [CrossRef]
- Sheehan, P.; Liapis, A.I. Modeling of the primary and secondary drying stages of the freeze drying of pharmaceutical products in vials: Numerical results obtained from the solution of a dynamic and spatially multi-dimensional lyophilization model for different operational policies. Biotechnol. Bioeng. 1998, 60, 712–728. [Google Scholar] [CrossRef]
- Srinivasan, G.; Muneeshwaran, M.; Raja, B. Numerical investigation of heat and mass transfer behavior of freeze drying of milk in vial. Heat Mass Transf. 2019, 55, 2073–2081. [Google Scholar] [CrossRef]
- Velardi, S.A.; Barresi, A.A. Development of simplified models for the freeze-drying process and investigation of the optimal operating conditions. Chem. Eng. Res. Des. 2008, 86, 9–22. [Google Scholar] [CrossRef]
- Vilas, C.; Alonso, A.A.; Balsa-Canto, E.; López-Quiroga, E.; Trelea, I.C. Model-Based Real Time Operation of the Freeze-Drying Process. Processes 2020, 8, 325. [Google Scholar] [CrossRef] [Green Version]
- Fissore, D.; Barresi, A.A. Scale-up and Process Transfer of Freeze-Drying Recipes. Dry. Technol. 2011, 29, 1673–1684. [Google Scholar] [CrossRef] [Green Version]
- Fissore, D.; Pisano, R. Computer-Aided Framework for the Design of Freeze-Drying Cycles: Optimization of the Operating Conditions of the Primary Drying Stage. Processes 2015, 3, 406–421. [Google Scholar] [CrossRef]
- Fissore, D.; Pisano, R.; Barresi, A.A. Advanced approach to build the design space for the primary drying of a pharmaceutical freeze-drying process. J. Pharm. Sci. 2011, 100, 4922–4933. [Google Scholar] [CrossRef]
- Koganti, V.R.; Shalaev, E.Y.; Berry, M.R.; Osterberg, T.; Youssef, M.; Hiebert, D.N.; Kanka, F.A.; Nolan, M.; Barrett, R.; Scalzo, G.; et al. Investigation of design space for freeze-drying: Use of modeling for primary drying segment of a freeze-drying cycle. AAPS PharmSciTech 2011, 12, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leys, L.; Vanbillemont, B.; van Bockstal, P.J.; Lammens, J.; Nuytten, G.; Corver, J.; Vervaet, C.; de Beer, T. A primary drying model-based comparison of conventional batch freeze-drying to continuous spin-freeze-drying for unit doses. Eur. J. Pharm. Biopharm. 2020, 157, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Shivkumar, G.; Kazarin, P.S.; Strongrich, A.D.; Alexeenko, A.A. LyoPRONTO: An Open-Source Lyophilization Process Optimization Tool. AAPS PharmSciTech 2019, 20, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanbillemont, B.; Nicolaï, N.; Leys, L.; de Beer, T. Model-Based Optimisation and Control Strategy for the Primary Drying Phase of a Lyophilisation Process. Pharmaceutics 2020, 12, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Shang, S.; Tharp, T.; Jameel, F.; Sinha, K.; Nere, N.K. Leveraging Lyophilization Modeling for Reliable Development, Scale-up and Technology Transfer. AAPS PharmSciTech 2019, 20, 263. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Moussa, E.M.; Witting, M.; Zhou, D.; Sinha, K.; Hirth, M.; Gastens, M.; Shang, S.; Nere, N.; Somashekar, S.C.; et al. Predictive models of lyophilization process for development, scale-up/tech transfer and manufacturing. Eur. J. Pharm. Biopharm. 2018, 128, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Arsiccio, A.; Pisano, R. Application of the Quality by Design Approach to the Freezing Step of Freeze-Drying: Building the Design Space. J. Pharm. Sci. 2018, 107, 1586–1596. [Google Scholar] [CrossRef]
- Jameel, F.; Hershenson, S.; Khan, M.A.; Martin-Moe, S. (Eds.) Quality by Design for Biopharmaceutical Drug Product Development; Springer: New York, NY, USA, 2015; ISBN 978-1-4939-2315-1. [Google Scholar]
- Mockus, L.N.; Paul, T.W.; Pease, N.A.; Harper, N.J.; Basu, P.K.; Oslos, E.A.; Sacha, G.A.; Kuu, W.Y.; Hardwick, L.M.; Karty, J.J.; et al. Quality by design in formulation and process development for a freeze-dried, small molecule parenteral product: A case study. Pharm. Dev. Technol. 2011, 16, 549–576. [Google Scholar] [CrossRef]
- Pisano, R.; Fissore, D.; Barresi, A.A.; Brayard, P.; Chouvenc, P.; Woinet, B. Quality by design: Optimization of a freeze-drying cycle via design space in case of heterogeneous drying behavior and influence of the freezing protocol. Pharm. Dev. Technol. 2013, 18, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Sixt, M.; Uhlenbrock, L.; Strube, J. Toward a Distinct and Quantitative Validation Method for Predictive Process Modelling—On the Example of Solid-Liquid Extraction Processes of Complex Plant Extracts. Processes 2018, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Wagner, W.; Riethmann, T.; Feistel, R.; Harvey, A.H. New Equations for the Sublimation Pressure and Melting Pressure of H2O Ice Ih. J. Phys. Chem. Ref. Data 2011, 40, 43103. [Google Scholar] [CrossRef] [Green Version]
- Giordano, A.; Barresi, A.A.; Fissore, D. On the use of mathematical models to build the design space for the primary drying phase of a pharmaceutical lyophilization process. J. Pharm. Sci. 2011, 100, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, L.A.; Ferris, S.J.; Nail, S.L. Experimental Aspects of Measuring the Vial Heat Transfer Coefficient in Pharmaceutical Freeze-Drying. AAPS PharmSciTech 2018, 19, 1810–1817. [Google Scholar] [CrossRef]
- Gieseler, H.; Kessler, W.J.; Finson, M.; Davis, S.J.; Mulhall, P.A.; Bons, V.; Debo, D.J.; Pikal, M.J. Evaluation of tunable diode laser absorption spectroscopy for in-process water vapor mass flux measurements during freeze drying. J. Pharm. Sci. 2007, 96, 1776–1793. [Google Scholar] [CrossRef] [PubMed]
- Kuu, W.Y.; Nail, S.L. Rapid freeze-drying cycle optimization using computer programs developed based on heat and mass transfer models and facilitated by tunable diode laser absorption spectroscopy (TDLAS). J. Pharm. Sci. 2009, 98, 3469–3482. [Google Scholar] [CrossRef]
- Milton, N.; Pikal, M.J.; Roy, M.L.; Nail, S.L. Evaluation of manometric temperature measurement as a method of monitoring product temperature during lyophilization. PDA J. Pharm. Sci. Technol. 1997, 51, 7–16. [Google Scholar]
- Tang, X.C.; Nail, S.L.; Pikal, M.J. Evaluation of manometric temperature measurement (MTM), a process analytical technology tool in freeze drying, part III: Heat and mass transfer measurement. AAPS PharmSciTech 2006, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Liapis, A.I.; Sadikoglu, H. Dynamic pressure rise in the drying chamber as a remote sensing method for monitoring the temperature of the product during the primary drying stage of freeze drying. Dry. Technol. 1998, 16, 1153–1171. [Google Scholar] [CrossRef]
- Kuu, W.Y.; O’Bryan, K.R.; Hardwick, L.M.; Paul, T.W. Product mass transfer resistance directly determined during freeze-drying cycle runs using tunable diode laser absorption spectroscopy (TDLAS) and pore diffusion model. Pharm. Dev. Technol. 2011, 16, 343–357. [Google Scholar] [CrossRef]
- Tang, X.C.; Nail, S.L.; Pikal, M.J. Evaluation of manometric temperature measurement, a process analytical technology tool for freeze-drying: Part II measurement of dry-layer resistance. AAPS PharmSciTech 2006, 7, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambhatla, S.; Ramot, R.; Bhugra, C.; Pikal, M.J. Heat and mass transfer scale-up issues during freeze drying: II. Control and characterization of the degree of supercooling. AAPS PharmSciTech 2004, 5, e58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuu, W.Y.; Hardwick, L.M.; Akers, M.J. Rapid determination of dry layer mass transfer resistance for various pharmaceutical formulations during primary drying using product temperature profiles. Int. J. Pharm. 2006, 313, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Rajniak, P.; Moreira, J.; Tsinontides, S.; Pham, D.; Bermingham, S. Integrated use of mechanistic models and targeted experiments for development, scale-up and optimization of lyophilization cycles: A single vial approach for primary drying. Dry. Technol. 2020, 1–16. [Google Scholar] [CrossRef]
- Barresi, A.A.; Pisano, R.; Fissore, D.; Rasetto, V.; Velardi, S.A.; Vallan, A.; Parvis, M.; Galan, M. Monitoring of the primary drying of a lyophilization process in vials. Chem. Eng. Process. Process Intensif. 2009, 48, 408–423. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.M.; Pikal, M.J. Lyophilization process design space. J. Pharm. Sci. 2013, 102, 3883–3887. [Google Scholar] [CrossRef] [PubMed]
- Schneid, S. Investigation of Novel Process Analytical Technology (PAT) Tools for Use in Freeze-Drying Processes. Ph.D. Thesis, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Erlangen, Germany, 2010. [Google Scholar]
- Patel, S.M.; Pikal, M. Process analytical technologies (PAT) in freeze-drying of parenteral products. Pharm. Dev. Technol. 2009, 14, 567–587. [Google Scholar] [CrossRef]
- Kauppinnen, A. Raman and Near-Infrared Spectroscopic Methods for In-Line Monitoring of Freeze-Drying Process. Ph.D. Thesis, University of Eastern Finland, Kuopio, Finland, 2015. [Google Scholar]
- De Beer, T.; Burggraeve, A.; Fonteyne, M.; Saerens, L.; Remon, J.P.; Vervaet, C. Near infrared and Raman spectroscopy for the in-process monitoring of pharmaceutical production processes. Int. J. Pharm. 2011, 417, 32–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brülls, M.; Folestad, S.; Sparén, A.; Rasmuson, A. In-situ near-infrared spectroscopy monitoring of the lyophilization process. Pharm. Res. 2003, 20, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Mensink, M.A.; van Bockstal, P.-J.; Pieters, S.; de Meyer, L.; Frijlink, H.W.; van der Voort Maarschalk, K.; Hinrichs, W.L.J.; de Beer, T. In-line near infrared spectroscopy during freeze-drying as a tool to measure efficiency of hydrogen bond formation between protein and sugar, predictive of protein storage stability. Int. J. Pharm. 2015, 496, 792–800. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, A.; Toiviainen, M.; Korhonen, O.; Aaltonen, J.; Järvinen, K.; Paaso, J.; Juuti, M.; Ketolainen, J. In-line multipoint near-infrared spectroscopy for moisture content quantification during freeze-drying. Anal. Chem. 2013, 85, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- De Beer, T.R.M.; Allesø, M.; Goethals, F.; Coppens, A.; Heyden, Y.V.; de Diego, H.L.; Rantanen, J.; Verpoort, F.; Vervaet, C.; Remon, J.P.; et al. Implementation of a process analytical technology system in a freeze-drying process using Raman spectroscopy for in-line process monitoring. Anal. Chem. 2007, 79, 7992–8003. [Google Scholar] [CrossRef]
- De Beer, T.R.M.; Vercruysse, P.; Burggraeve, A.; Quinten, T.; Ouyang, J.; Zhang, X.; Vervaet, C.; Remon, J.P.; Baeyens, W.R.G. In-line and real-time process monitoring of a freeze drying process using Raman and NIR spectroscopy as complementary process analytical technology (PAT) tools. J. Pharm. Sci. 2009, 98, 3430–3446. [Google Scholar] [CrossRef]
- Tang, X.C.; Nail, S.L.; Pikal, M.J. Freeze-drying process design by manometric temperature measurement: Design of a smart freeze-dryer. Pharm Res 2005, 22, 685–700. [Google Scholar] [CrossRef]
- Fissore, D.; Pisano, R.; Barresi, A.A. On the Methods Based on the Pressure Rise Test for Monitoring a Freeze-Drying Process. Dry. Technol. 2010, 29, 73–90. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.I.; Akers, M.J.; Nail, S.L. The physical state of mannitol after freeze-drying: Effects of mannitol concentration, freezing rate, and a noncrystallizing cosolute. J. Pharm. Sci. 1998, 87, 931–935. [Google Scholar] [CrossRef]
- Patel, S.M.; Jameel, F.; Pikal, M.J. The effect of dryer load on freeze drying process design. J. Pharm. Sci. 2010, 99, 4363–4379. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
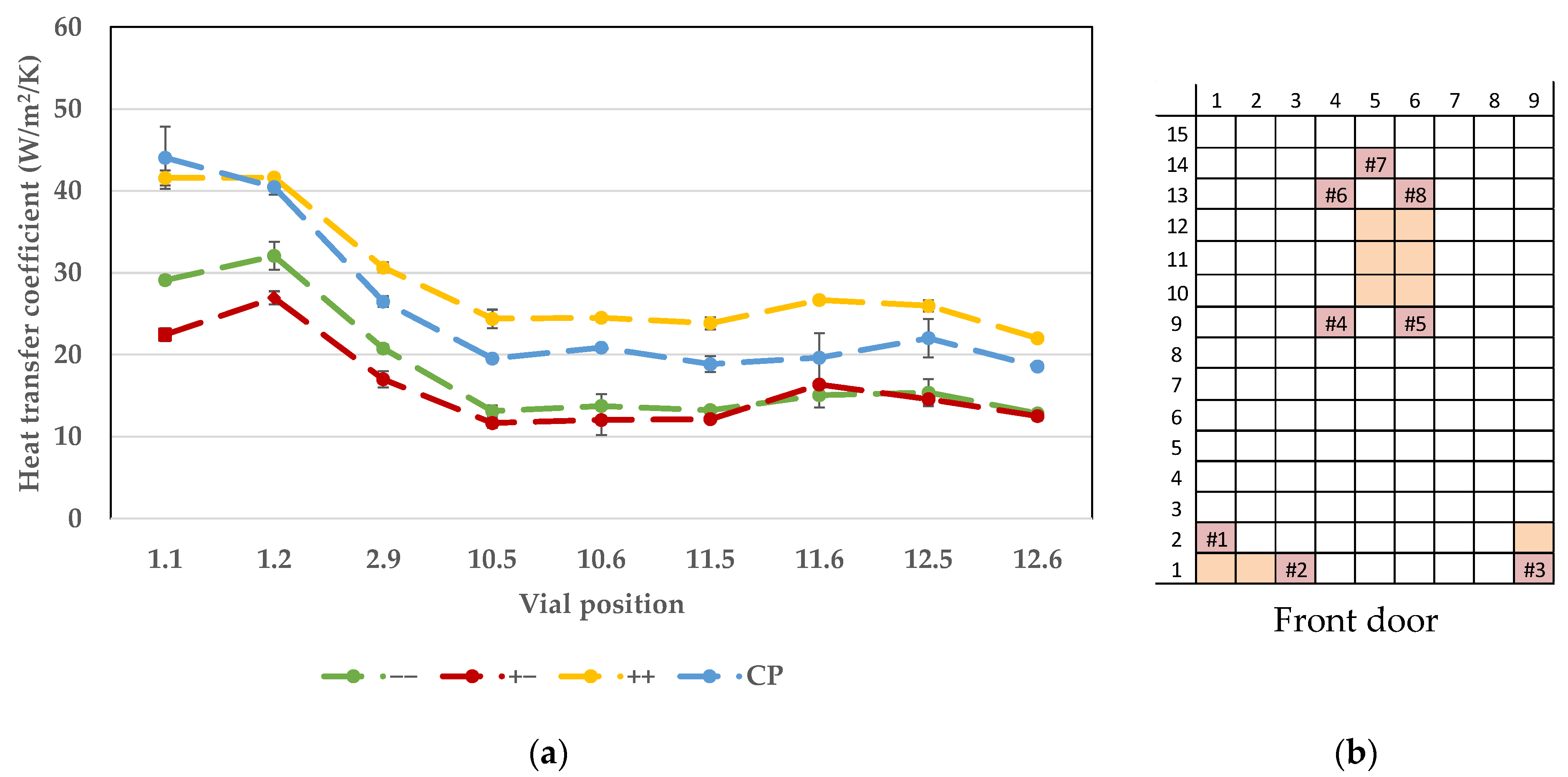{kind=link}
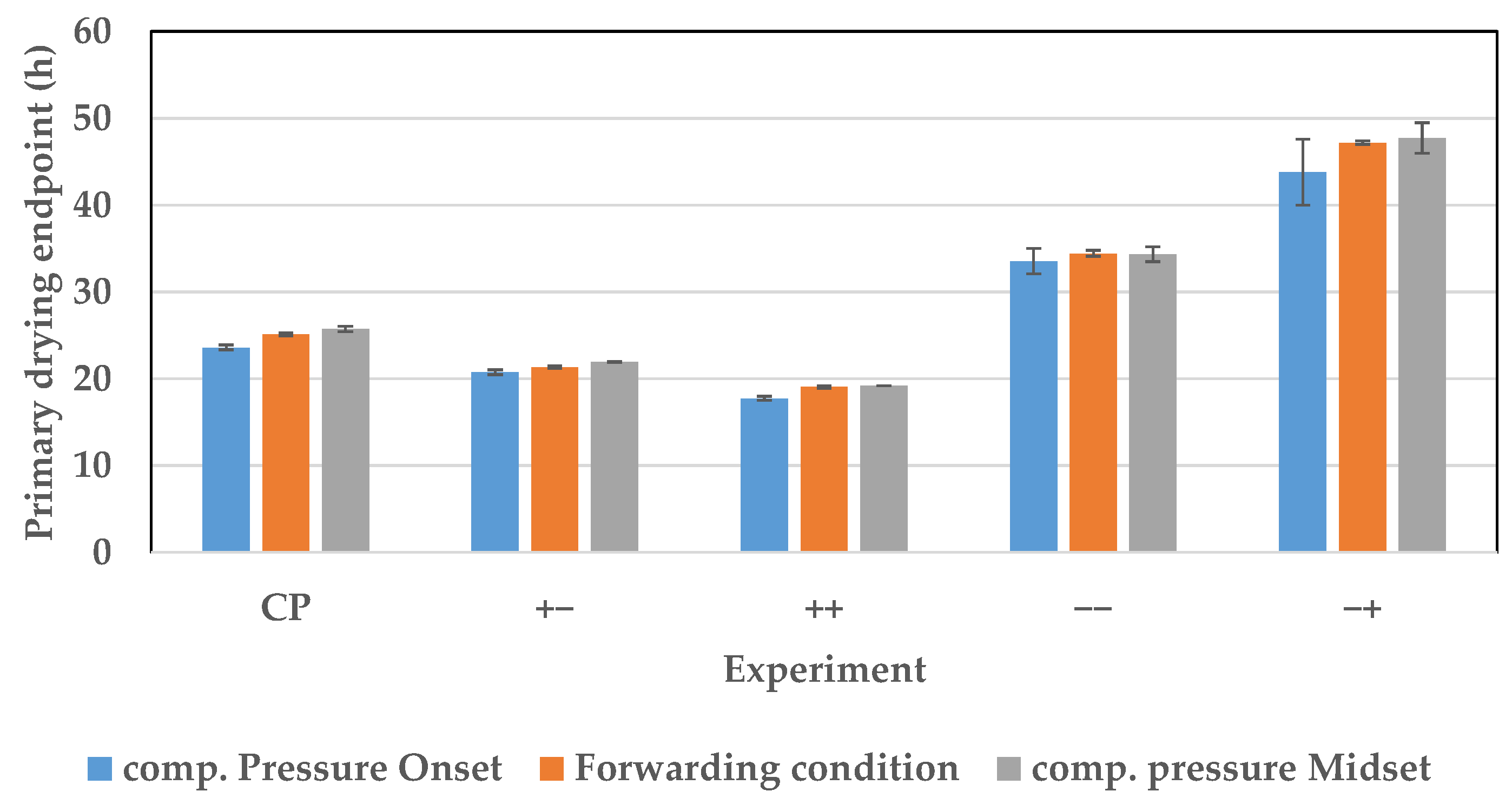{kind=link}
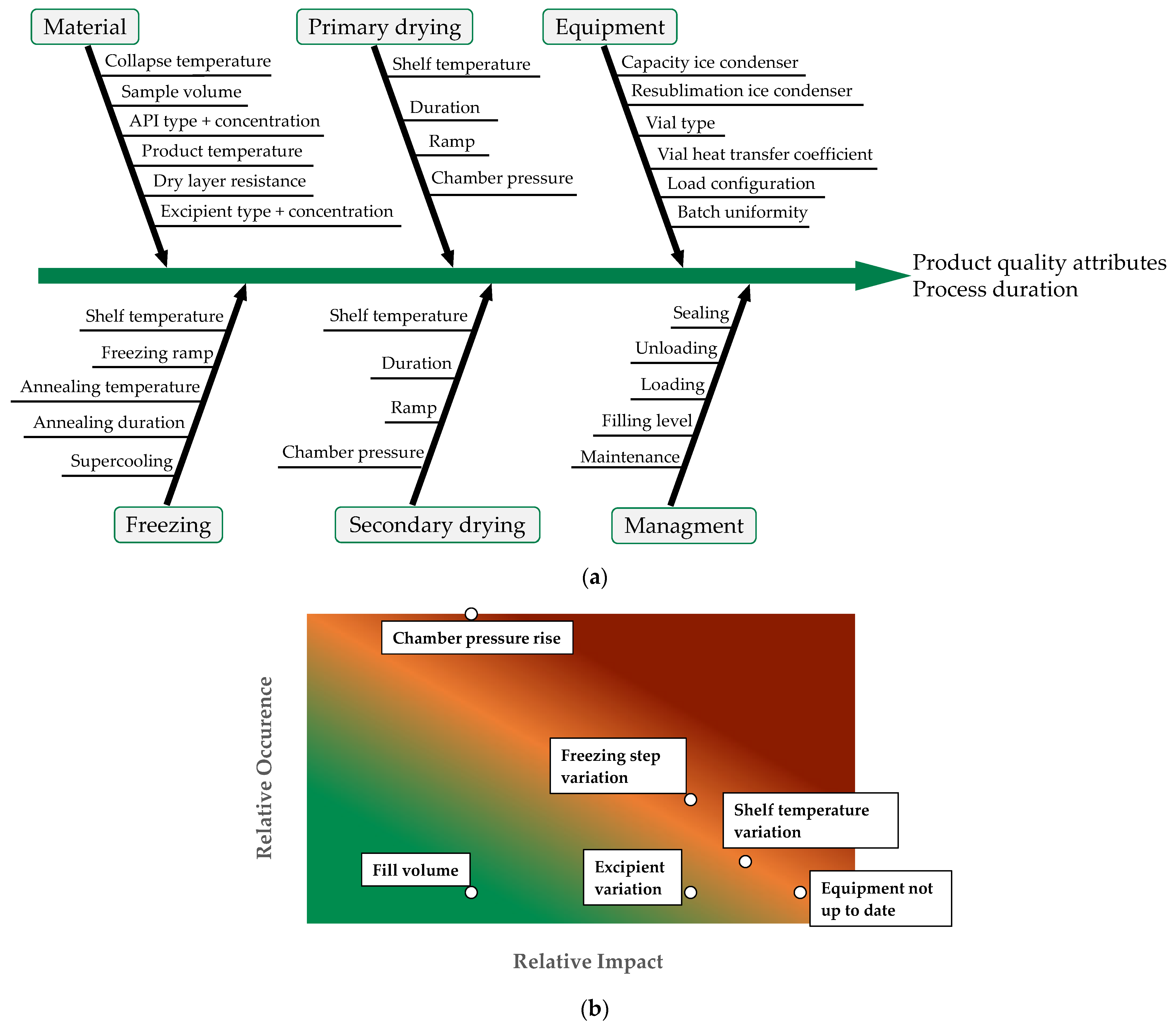
| Risk | Impact | Occurrence | Comment |
|---|---|---|---|
| Shelf temperature variation | 8 | 2 | Shelf temperature affects drying rate in both phases, too high value could cause collapse but a low value leads to long drying times, once freeze drying recipe is set, temperature easy to control |
| Chamber pressure rise | 3 | 10 | High pressure could lead to melt back, during MTM pressure is increased in an interval that is safe for the product |
| Freezing step variation | 7 | 4 | Freezing step sets the foundation for drying phases, once freezing recipe is set the temperature can be controlled easily but stochastic nature of nucleation still leads to small deviations in ice crystal size |
| Equipment not up to date | 9 | 1 | Uncalibrated measurement units lead deviation from recipe, prevention through maintenance intervals |
| Excipient variation | 7 | 1 | Formulation has high impact because it sets the failure mode, once formulation is set, composition can be good controlled |
| Fill level | 3 | 1 | Increased height can lead to collapse if the process is rigorously optimized, but fill level can be controlled easily |
| # | Primary Drying | ||
|---|---|---|---|
| Shelf Temperature (°C) | Chamber Pressure (mbar) | ||
| 1 | +− | −15 | 0.133 |
| 2 | ++ | −15 | 0.57 |
| 3 | −− | −25 | 0.133 |
| 4 | +− | −15 | 0.133 |
| 5 | ++ | −15 | 0.57 |
| 6 | −− | −25 | 0.133 |
| 7 | −+ | −25 | 0.57 |
| 8 | −+ | −25 | 0.57 |
| 9 | CP | −20 | 0.352 |
| 10 | CP | −20 | 0.352 |
| 11 | CP | −20 | 0.352 |
| Parameter | Value |
|---|---|
| R1 (m/s) | 50,555.24 ± 5895.69 |
| R2 (1/s) | 2.06 × 107 ± 1.14 × 107 |
| R3 (1/m) | 147.41 ± 214.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juckers, A.; Knerr, P.; Harms, F.; Strube, J. Advanced Process Analytical Technology in Combination with Process Modeling for Endpoint and Model Parameter Determination in Lyophilization Process Design and Optimization. Processes 2021, 9, 1600. https://doi.org/10.3390/pr9091600
Juckers A, Knerr P, Harms F, Strube J. Advanced Process Analytical Technology in Combination with Process Modeling for Endpoint and Model Parameter Determination in Lyophilization Process Design and Optimization. Processes. 2021; 9(9):1600. https://doi.org/10.3390/pr9091600
Chicago/Turabian StyleJuckers, Alex, Petra Knerr, Frank Harms, and Jochen Strube. 2021. "Advanced Process Analytical Technology in Combination with Process Modeling for Endpoint and Model Parameter Determination in Lyophilization Process Design and Optimization" Processes 9, no. 9: 1600. https://doi.org/10.3390/pr9091600