
Fast and Flexible mRNA Vaccine Manufacturing as a Solution to Pandemic Situations by Adopting Chemical Engineering Good Practice—Continuous Autonomous Operation in Stainless Steel Equipment Concepts




Abstract
:1. Introduction
2. Materials and Methods
2.1. Model Overview
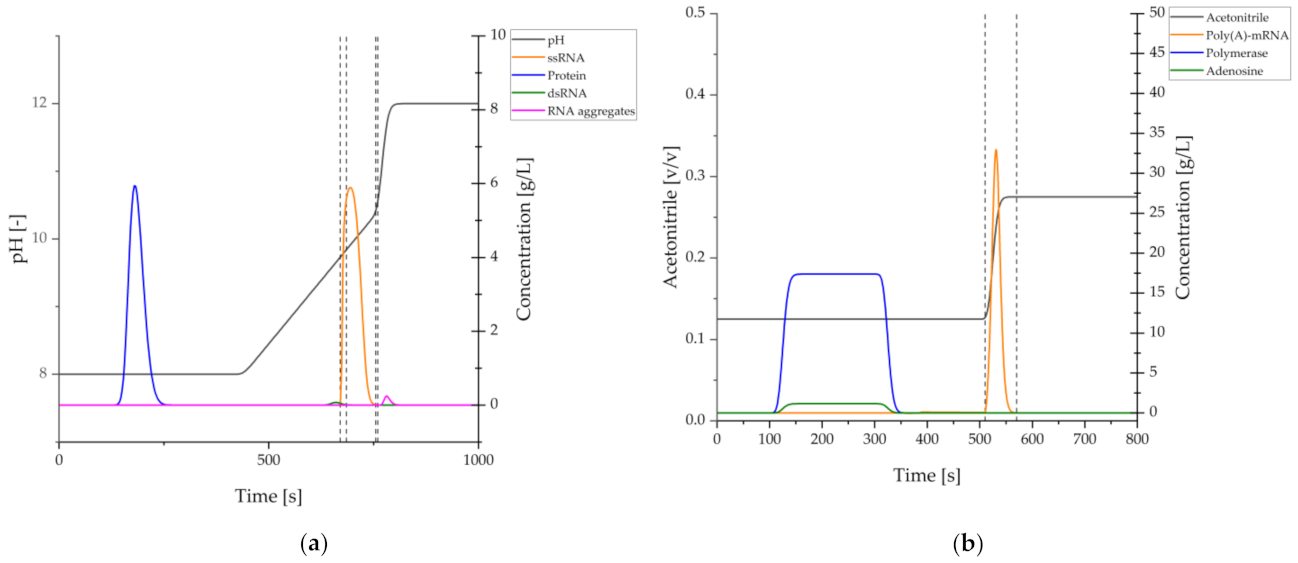
2.1.1. Adsorptive Purification Processes
2.1.2. Continuous Adsorptive Processes
2.2. Cost Estimation
3. Results
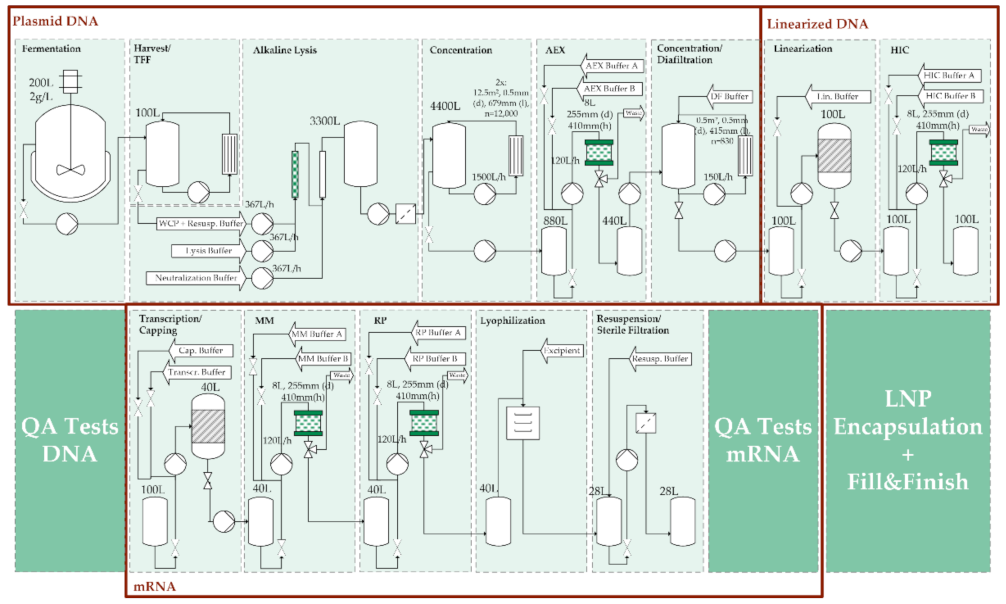
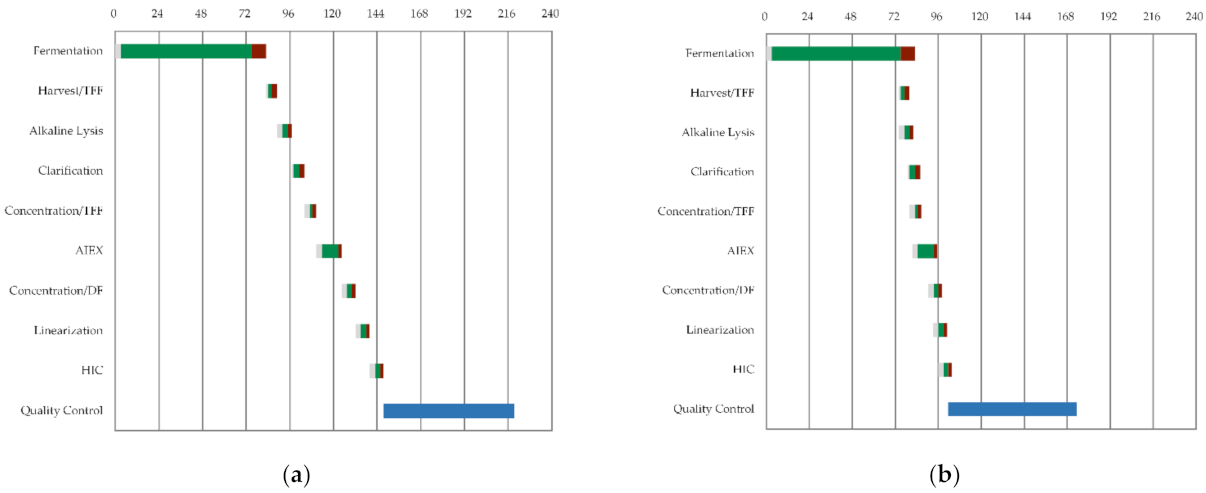
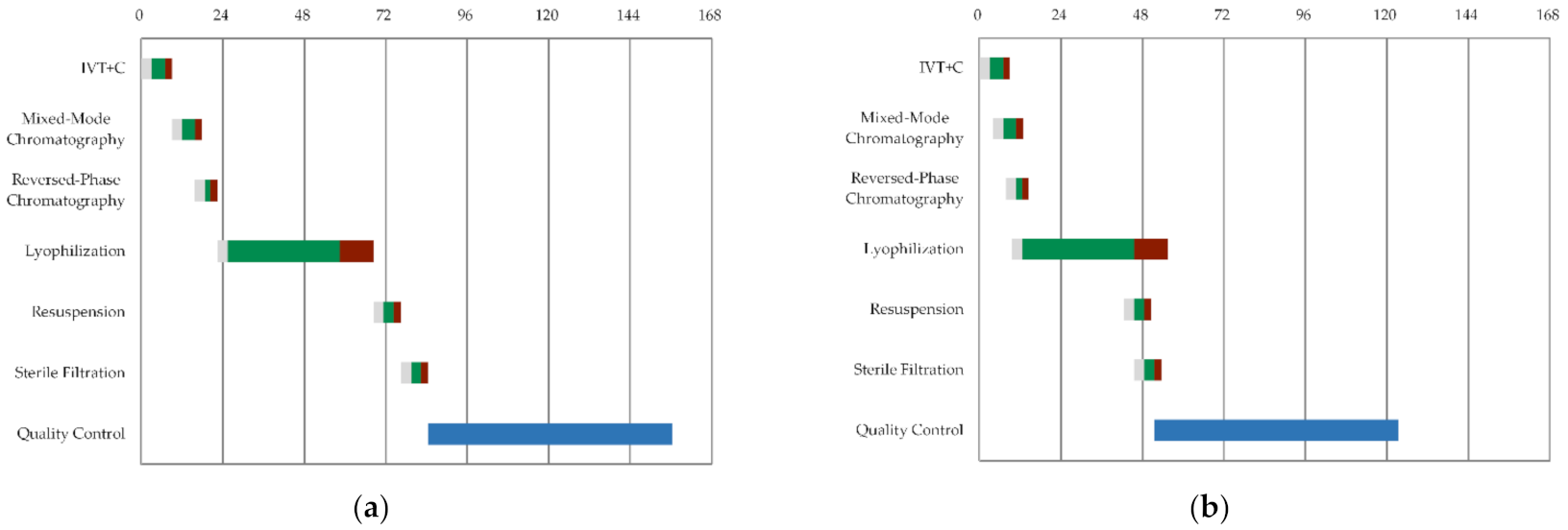
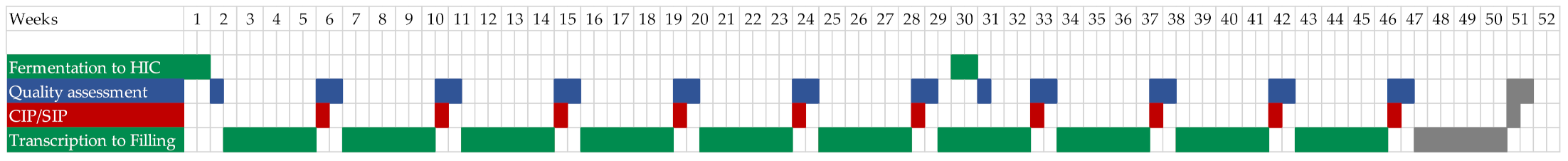
3.1. General Process Design and Schedule Scenarios
CAPEX/OPEX
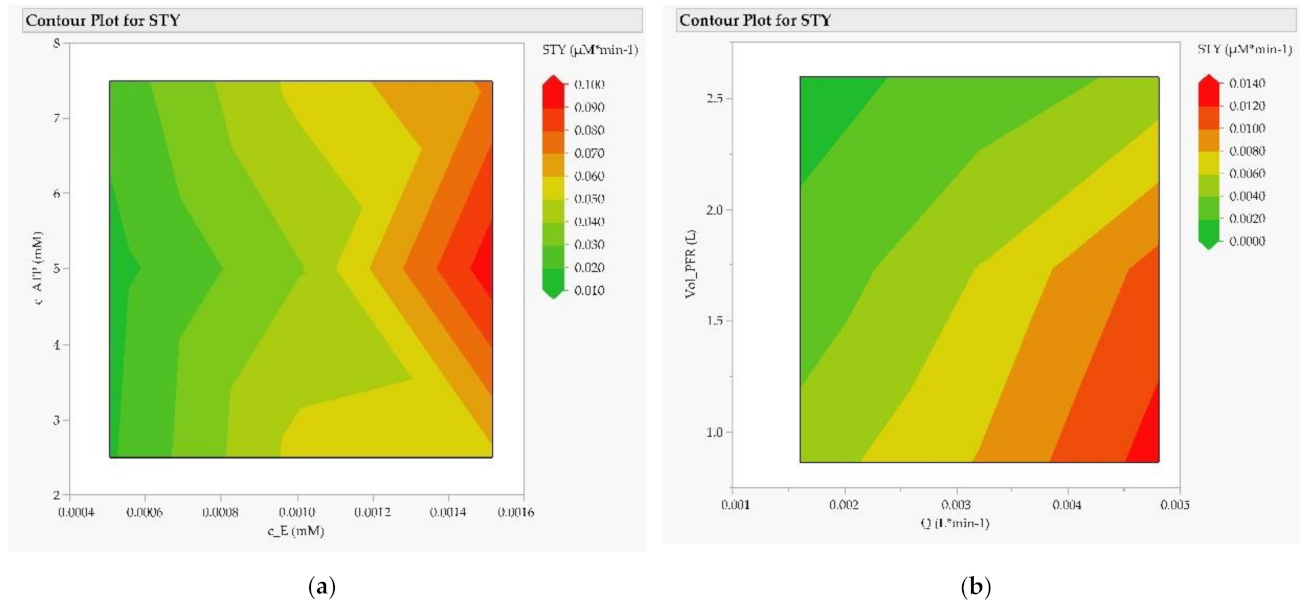
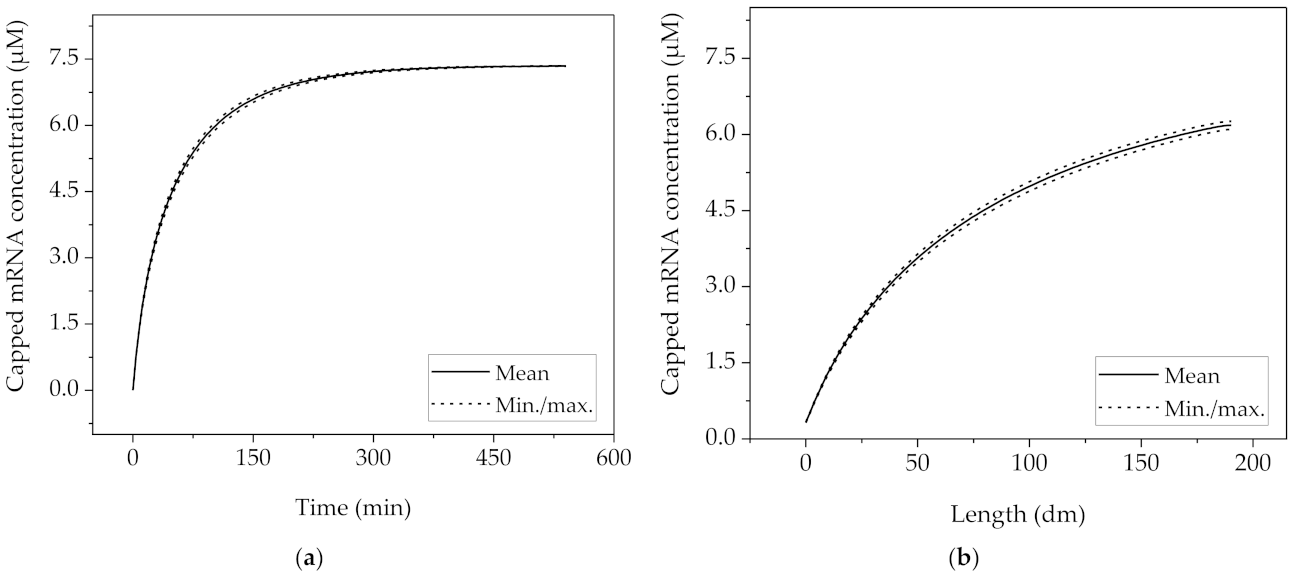
3.2. Purification of mRNA with Mixed-Mode Chromatography
3.3. Purification of Poly-mRNA with Reversed-Phase Chromatography
3.3.1. Continuous Processing Using Monoliths
3.3.2. Bead-Based Solutions
4. Discussion
4.1. Critical Raw Materials
4.2. Manufacturing Mode
4.3. Equipment Technology
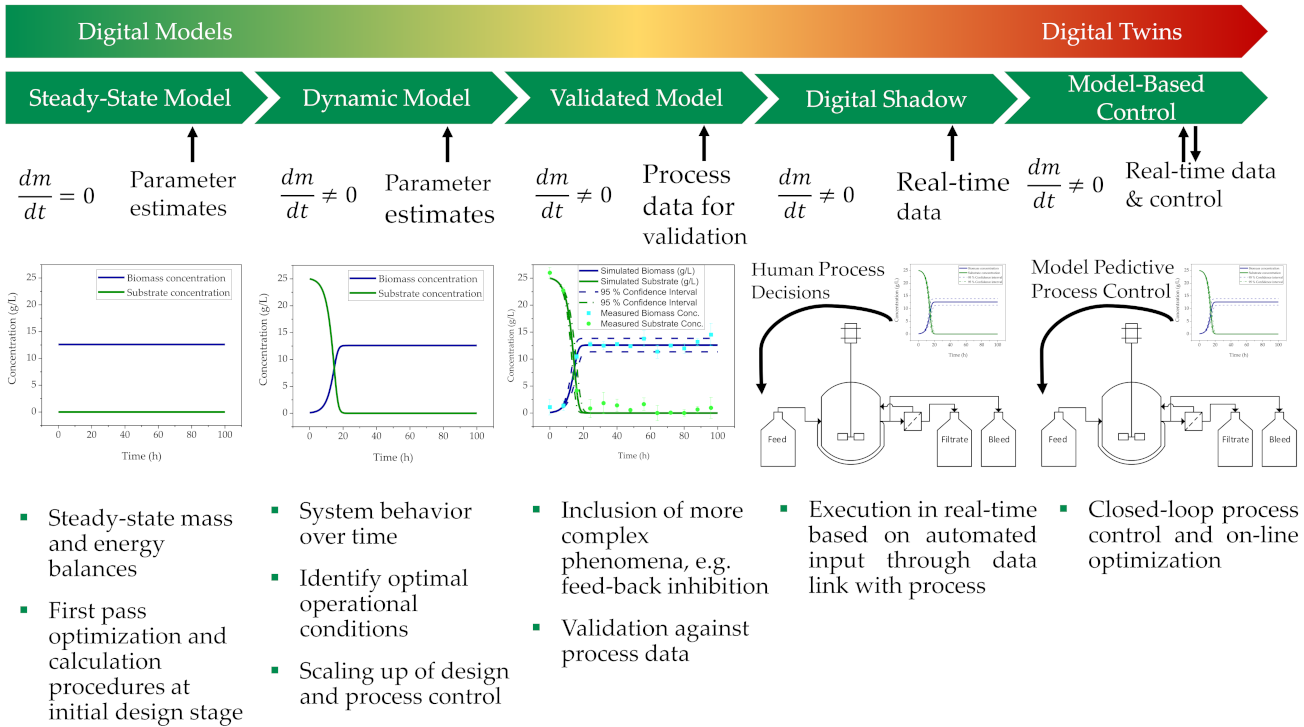
4.4. Process Control
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- European Medicines Agency. COVID-19: How EMA Fast-Tracks Development Support and Approval of Medicines and Vaccines. Press Release. Available online: https://www.ema.europa.eu/en/news/covid-19-how-ema-fast-tracks-development-support-approval-medicines-vaccines (accessed on 24 September 2021).
- FDA. Emergency Use Authorization for Vaccines Explained. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/emergency-use-authorization-vaccines-explained (accessed on 24 September 2021).
- Davis, N. How Has a Covid Vaccine Been Developed So Quickly? Analysis: Funding and High Public Interest Contributed to Slashing of Research and Approval Time. Available online: https://www.theguardian.com/society/2020/dec/08/how-has-a-covid-vaccine-been-developed-so-quickly (accessed on 24 September 2021).
- Bill & Melinda Gates Foundation. Bill & Melinda Gates Foundation, Wellcome, and Mastercard Launch Initiative to Speed Development and Access to Therapies for COVID-19: Press Release. Available online: https://www.gatesfoundation.org/Media-Center/Press-Releases/2020/03/COVID-19-Therapeutics-Accelerator (accessed on 11 March 2021).
- Brors, P.; Hofmann, S. Dietmar Hopp Will Mit Curevac “Rennen um Besten Impfstoff Gewinnen”. Available online: https://www.handelsblatt.com/unternehmen/management/der-risikoinvestor-dietmar-hopp-will-mit-curevac-rennen-um-besten-impfstoff-gewinnen/26154156.html?ticket=ST-13985126-bIPozoMDRzkCluynaUe7-ap3 (accessed on 11 March 2021).
- Wikipedia. Andreas und Thomas Strüngmann. Available online: https://de.wikipedia.org/wiki/Andreas_und_Thomas_Str%C3%BCngmann (accessed on 24 September 2021).
- Beaumont, P. How Are Covid Vaccines Produced and Why Have There Been Delays? The Newness of Some of the Technology and Gaps in Global Preparedness Have Led to Bottlenecks. Available online: https://www.theguardian.com/society/2021/mar/19/how-are-covid-vaccines-produced-and-why-have-there-been-delays (accessed on 24 September 2021).
- Lupkin, S. The U.S. Paid Billions To Get Enough COVID Vaccines Last Fall. What Went Wrong? Available online: https://www.npr.org/sections/health-shots/2021/08/25/1029715721/pfizer-vaccine-operation-warp-speed-delay?t=1632300860357 (accessed on 24 September 2021).
- Tapper, J. Global Covid Vaccine Rollout Threatened by Shortage of Vital Components: Pharmaceutical Firms Warn of Delays to Items Such as the Large Bags in Which Vaccine Cells Are Grown. Available online: https://www.theguardian.com/world/2021/apr/10/global-covid-vaccine-rollout-threatened-by-shortage-of-vital-components (accessed on 24 September 2021).
- Barnhill, C. The COVID-19 Vaccine Supply Chain: Potential Problems and Bottlenecks. Available online: https://scm.ncsu.edu/scm-articles/article/the-covid-19-vaccine-supply-chain-potential-problems-and-bottlenecks (accessed on 24 September 2021).
- WTO. Indicative List of Trade-Related Bottlenecks and Trade-Facilitating Measures on Critical Products to Combat Covid-19: Information Note. Available online: https://www.wto.org/english/tratop_e/covid19_e/bottlenecks_report_e.pdf (accessed on 24 September 2021).
- Kansteiner, F. The Next Big COVID-19 Bottleneck? A Shortage of Trained Vaccine Workers, Experts Say. Available online: https://www.fiercepharma.com/manufacturing/next-big-covid-19-bottleneck-a-shortage-trained-vaccine-workers-pharmas-and (accessed on 24 September 2021).
- Samsung Biologics. Economics of Stainless Steel and Single-Use Systems. Available online: https://samsungbiologics.com/media/science-technology-view?boardSeq=989&schBoardCtgryCcd=&schString=&schBoardYear=&boardDtm=1607353200000&page=2 (accessed on 24 September 2021).
- Samsung Biologics. Key Considerations in Choosing Stainless Steel vs. Single Use Bioprocessing. Available online: https://samsungbiologics.com/media/science-technology-view?boardSeq=1254&schBoardCtgryCcd=&schString=&schBoardYear=&boardDtm=1624806000000&page=1 (accessed on 24 September 2021).
- Sommerfeld, S.; Strube, J. Challenges in biotechnology production—Generic processes and process optimization for monoclonal antibodies. Chem. Eng. Process. Process. Intensif. 2005, 44, 1123–1137. [Google Scholar] [CrossRef]
- Strube, J.; Sommerfeld, S.; Lohrmann, M. Process Development and Optimization for Biotechnology Production—Monoclonal Antibodies. In Bioseparation and Bioprocessing, 2nd ed.; Subramanian, G., Ed.; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Biospace. New capacity Helps Eliminate Bottleneck for COVID-19 Vaccines. Available online: https://www.biospace.com/article/releases/new-capacity-helps-eliminate-bottleneck-for-covid-19-vaccines/ (accessed on 24 September 2021).
- Christensen, J. Quality Issue at Baltimore Vaccine Plant Delays Some of Johnson & Johnson’s Vaccine. Available online: https://edition.cnn.com/2021/03/31/health/johnson--johnson-vaccine-manufacturing-problem/index.html (accessed on 24 September 2021).
- Rees, V. EDQM Releases New Guidelines for COVID-19 Vaccine Quality Testing. Available online: https://www.europeanpharmaceuticalreview.com/news/133928/edqm-releases-new-guidelines-for-covid-19-vaccine-quality-testing/ (accessed on 24 September 2021).
- Schmidt, A.; Strube, J. Distinct and Quantitative Validation Method for Predictive Process Modeling with Examples of Liquid-Liquid Extraction Processes of Complex Feed Mixtures. Processes 2019, 7, 298. [Google Scholar] [CrossRef] [Green Version]
- Uhlenbrock, L.; Sixt, M.; Strube, J. Quality-by-Design (QbD) process evaluation for phytopharmaceuticals on the example of 10-deacetylbaccatin III from yew. Resour.-Effic. Technol. 2017, 3, 137–143. [Google Scholar] [CrossRef]
- Helgers, H.; Hengelbrock, A.; Schmidt, A.; Strube, J. Digital Twin for Continuous mRNA Production: Submitted; MDPI: Basel, Switzerland, 2021. [Google Scholar]
- Udugama, I.A.; Lopez, P.C.; Gargalo, C.L.; Li, X.; Bayer, C.; Gernaey, K.V. Digital Twin in biomanufacturing: Challenges and opportunities towards its implementation. Syst. Microbiol. Biomanuf. 2021, 1, 257–274. [Google Scholar] [CrossRef]
- Schmidt, A.; Helgers, H.; Vetter, F.L.; Juckers, A.; Strube, J. Digital Twin of mRNA-Based SARS-COVID-19 Vaccine Manufacturing towards Autonomous Operation for Improvements in Speed, Scale, Robustness, Flexibility and Real-Time Release Testing. Processes 2021, 9, 748. [Google Scholar] [CrossRef]
- Guiochon, G. Fundamentals of Preparative and Nonlinear Chromatography, 2nd ed.; Elsevier Acad. Press: Amsterdam, The Netherlands, 2006; ISBN 9780123705372. [Google Scholar]
- Halan, V.; Maity, S.; Bhambure, R.; Rathore, A.S. Multimodal Chromatography for Purification of Biotherapeutics—A Review. Curr. Protein Pept. Sci. 2019, 20, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Matos, T.; Queiroz, J.A.; Bülow, L. Plasmid DNA purification using a multimodal chromatography resin. J. Mol. Recognit. 2014, 27, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Kanavarioti, A. HPLC methods for purity evaluation of man-made single-stranded RNAs. Sci. Rep. 2019, 9, 1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Santos, A.R.; Alves, C.P.; Monteiro, G.; Azevedo, A.M.; Prazeres, D.M.F. Multimodal chromatography of supercoiled minicircles: A closer look into DNA-ligand interactions. Sep. Purif. Technol. 2019, 212, 161–170. [Google Scholar] [CrossRef]
- Dickman, M.J. Ion Pair Reverse-Phase Chromatography: A Versatile Platform for the Analysis of RNA; Chromatography Today: St. Albans, UK, 2011. [Google Scholar]
- Levine, A.; Ono, T.; Hirose, T. HPLC Purification of mRNA with Reverse Phase and Size Exclusion Chromatography. 2019. Available online: https://www.nacalai.co.jp/global/download/pdf/TIDES_2019_poster_mrna.pdf (accessed on 24 September 2021).
- Seidel-Morgenstern, A. Experimental determination of single solute and competitive adsorption isotherms. J. Chromatogr. A 2004, 1037, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Zobel-Roos, S. Entwicklung, Modellierung und Validierung von Integrierten Kontinuierlichen Gegenstrom-Chromatographie-Prozessen. Ph.D. Thesis, Technische Universität Clausthal, Clausthal-Zellerfeld, Germany, 2018. [Google Scholar]
- Carta, G.; Rodrigues, A.E. Diffusion and convection in chromatographic processes using permeable supports with a bidisperse pore structure. Chem. Eng. Sci. 1993, 48, 3927–3935. [Google Scholar] [CrossRef]
- Wilson, E.J.; Geankoplis, C.J. Liquid Mass Transfer at Very Low Reynolds Numbers in Packed Beds. Ind. Eng. Chem. Fund. 1966, 5, 9–14. [Google Scholar] [CrossRef]
- Roberts, J.A.; Kimerer, L.; Carta, G. Effects of molecule size and resin structure on protein adsorption on multimodal anion exchange chromatography media. J. Chromatogr. A 2020, 1628, 461444. [Google Scholar] [CrossRef] [PubMed]
- SiliCycle. SiliaSphere™ Spherical Silica Gels. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwj1zZWdkvTxAhWS_rsIHRAhC1sQFjAAegQIBxAD&url=https%3A%2F%2Fwww.dichrom.com%2Fapp%2Fdownload%2F5805621860%2FSiliCycle-SiliaSphere-Brochure.pdf&usg=AOvVaw08hBaUHAqY3MNET7RJL3gx (accessed on 24 September 2021).
- Müller-Späth, T.; Aumann, L.; Melter, L.; Ströhlein, G.; Morbidelli, M. Chromatographic separation of three monoclonal antibody variants using multicolumn countercurrent solvent gradient purification (MCSGP). Biotechnol. Bioeng. 2008, 100, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Godawat, R.; Brower, K.; Jain, S.; Konstantinov, K.; Riske, F.; Warikoo, V. Periodic counter-current chromatography -- design and operational considerations for integrated and continuous purification of proteins. Biotechnol. J. 2012, 7, 1496–1508. [Google Scholar] [CrossRef]
- Strube, J. Technische Chromatographie: Auslegung, Optimierung, Betrieb und Wirtschaftlichkeit; TU Dortmund., Habilitation.: 1999; Shaker: Aachen, Germany, 2000; ISBN 3826568974. [Google Scholar]
- Zobel, S.; Helling, C.; Ditz, R.; Strube, J. Design and Operation of Continuous Countercurrent Chromatography in Biotechnological Production. Ind. Eng. Chem. Res. 2014, 53, 9169–9185. [Google Scholar] [CrossRef]
- Jungbauer, A. Continuous downstream processing of biopharmaceuticals. Trends Biotechnol. 2013, 31, 479–492. [Google Scholar] [CrossRef]
- Aumann, L.; Morbidelli, M. A continuous multicolumn countercurrent solvent gradient purification (MCSGP) process. Biotechnol. Bioeng. 2007, 98, 1043–1055. [Google Scholar] [CrossRef]
- Labarta, I.; Hoffman, S.; Simpkins, A. Manufacturing Strategy for the Production of 200 Million Sterile Doses of an mRNA Vaccine for COVID-19; Senior Design Reports; University of Pennsylvania: Philadelphia, PA, USA, 2021. [Google Scholar]
- Kis, Z.; Kontoravdi, C.; Shattock, R.; Shah, N. Resources, Production Scales and Time Required for Producing RNA Vaccines for the Global Pandemic Demand. Vaccines 2020, 9, 3. [Google Scholar] [CrossRef]
- Merck KGaA. Sigma Aldrich. Available online: https://www.sigmaaldrich.com/DE/de (accessed on 24 September 2021).
- VWR. Available online: https://de.vwr.com/store/ (accessed on 24 September 2021).
- Von der Mülbe, F.; Reidel, L.; Ketterer, T.; Gontcharova, L.; Pascolo, S.; Probst, J.; Schmid, A.; Bauer, S. Method for Producing RNA. U.S. Patent 10711315B2, 14 July 2020. [Google Scholar]
- Weise, E.; Weintraub, K. A COVID-19 Vaccine Life Cycle: From DNA to Doses. 2021. Available online: https://eu.usatoday.com/in-depth/news/health/2021/02/07/how-covid-vaccine-made-step-step-journey-pfizer-dose/4371693001/ (accessed on 6 March 2021).
- Peters, M.S.; Timmerhaus, K.D.; West, R.E. Plant Design and Economics for Chemical Engineers; McGraw-Hill: New York, NY, USA, 2003. [Google Scholar]
- Green, D.W.; Southard, M.Z. Perry’s Chemical Engineers’ Handbook; McGraw-Hill Education: New York, NY, USA, 2019; ISBN 0071834087. [Google Scholar]
- Abrams Kaplan, D.; Wehrwein, P. The Price Tags on the COVID-19 Vaccines. Available online: https://www.managedhealthcareexecutive.com/view/the-price-tags-on-the-covid-19-vaccines (accessed on 24 September 2021).
- Rosa, S.S.; Prazeres, D.M.F.; Azevedo, A.M.; Marques, M.P.C. mRNA vaccines manufacturing: Challenges and bottlenecks. Vaccine 2021, 39, 2190–2200. [Google Scholar] [CrossRef]
- BIA Separations. Purification of mRNA with CIMmultus PrimaS™: Technical Note. 2020. Available online: https://www.biaseparations.com/en/library/technical-notes/1088/purification-of-mrna-with-cimmultus-primastm (accessed on 24 September 2021).
- BIA Separations. Monolith Technology. Available online: https://www.biaseparations.com/en/featured/messenger-rna (accessed on 24 September 2021).
- Urbas, L. CIM Monolithic Columns for Purification and Analytics of Biomolecules. 2021. Available online: https://hp-ne.com/wp-content/uploads/2021/03/High_Purit-general_presentation.pdf (accessed on 24 September 2021).
- Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000; ISBN 3527306730.
- Kornecki, M.; Strube, J. Accelerating Biologics Manufacturing by Upstream Process Modelling. Processes 2019, 7, 166. [Google Scholar] [CrossRef] [Green Version]
- Sartorius AG. Preparing for the Emergence of mRNA Therapeutics. Available online: https://www.sartorius.com/en/applications/biopharmaceutical-manufacturing/mrna-production?utm_source=bing&utm_medium=cpc&utm_campaign=ww_en_search_BIA%20Application%20mRNA (accessed on 24 September 2021).
- Davies, J.G.; Gao, D.; Kim, Y.J.; Harris, R.; Cash, P.W.; Schofield, T.L.; Zhang, R.; Qin, Q. ICH Q5C Stability Testing of Biotechnological/Biological Products. In ICH Quality Guidelines: An Implementation Guide; Wiley: Hoboken, NJ, USA, 2017; p. 345. [Google Scholar]
- Kornecki, M.; Schmidt, A.; Strube, J. PAT as key-enabling technology for QbD in pharmaceutical manufacturing A conceptual review on upstream and downstream processing. Chim. Oggi-Chem. Today 2018, 36, 44–48. [Google Scholar]
- Zobel-Roos, S.; Mouellef, M.; Ditz, R.; Strube, J. Distinct and Quantitative Validation Method for Predictive Process Modelling in Preparative Chromatography of Synthetic and Bio-Based Feed Mixtures Following a Quality-by-Design (QbD) Approach. Processes 2019, 7, 580. [Google Scholar] [CrossRef] [Green Version]
- Zobel-Roos, S.; Schmidt, A.; Mestmäcker, F.; Mouellef, M.; Huter, M.; Uhlenbrock, L.; Kornecki, M.; Lohmann, L.; Ditz, R.; Strube, J. Accelerating Biologics Manufacturing by Modeling or: Is Approval under the QbD and PAT Approaches Demanded by Authorities Acceptable Without a Digital-Twin? Processes 2019, 7, 94. [Google Scholar] [CrossRef] [Green Version]
- Huter, M.J.; Strube, J. Model-Based Design and Process Optimization of Continuous Single Pass Tangential Flow Filtration Focusing on Continuous Bioprocessing. Processes 2019, 7, 317. [Google Scholar] [CrossRef] [Green Version]
- Roth, T.; Uhlenbrock, L.; Strube, J. Distinct and Quantitative Validation for Predictive Process Modelling in Steam Distillation of Caraway Fruits and Lavender Flower Following a Quality-By-Design (QbD) Approach. Processes 2020, 8, 594. [Google Scholar] [CrossRef]
- Zobel-Roos, S.; Mouellef, M.; Siemers, C.; Strube, J. Process Analytical Approach towards Quality Controlled Process Automation for the Downstream of Protein Mixtures by Inline Concentration Measurements Based on Ultraviolet/Visible Light (UV/VIS) Spectral Analysis. Antibodies 2017, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Barresi, A.A.; Fissore, D. In-Line Product Quality Control of Pharmaceuticals in Freeze-Drying Processes. In Modern Drying Technology; Tsotsas, E., Mujumdar, A.S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 91–154. ISBN 9783527631667. [Google Scholar]
- Pietrzykowski, M.; Flanagan, W.; Pizzi, V.; Brown, A.; Sinclair, A.; Monge, M. An environmental life cycle assessment comparison of single-use and conventional process technology for the production of monoclonal antibodies. J. Clean. Prod. 2013, 41, 150–162. [Google Scholar] [CrossRef]
- Schmidt, A.; Uhlenbrock, L.; Strube, J. Technical Potential for Energy and GWP Reduction in Chemical–Pharmaceutical Industry in Germany and EU—Focused on Biologics and Botanicals Manufacturing. Processes 2020, 8, 818. [Google Scholar] [CrossRef]
- Kornecki, M.; Schmidt, A.; Lohmann, L.; Huter, M.; Mestmäcker, F.; Klepzig, L.; Mouellef, M.; Zobel-Roos, S.; Strube, J. Accelerating Biomanufacturing by Modeling of Continuous Bioprocessing—Piloting Case Study of Monoclonal Antibody Manufacturing. Processes 2019, 7, 495. [Google Scholar] [CrossRef] [Green Version]
- Srai, J.S.; Badman, C.; Krumme, M.; Futran, M.; Johnston, C. Future Supply Chains Enabled by Continuous Processing—Opportunities and Challenges. May 20–21, 2014 Continuous Manufacturing Symposium. J. Pharm. Sci. 2015, 104, 840–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, G. Continuous Biomanufacturing—Innovative Technologies and Methods; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017; ISBN 9783527699902. [Google Scholar]
- Walther, J.; Godawat, R.; Hwang, C.; Abe, Y.; Sinclair, A.; Konstantinov, K. The business impact of an integrated continuous biomanufacturing platform for recombinant protein production. J. Biotechnol. 2015, 213, 3–12. [Google Scholar] [CrossRef] [PubMed]
- FDA. Guidance for Industry: PAT—A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance. 2004. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm070305.pdf (accessed on 19 February 2018).
- Feidl, F.; Vogg, S.; Wolf, M.; Podobnik, M.; Ruggeri, C.; Ulmer, N.; Wälchli, R.; Souquet, J.; Broly, H.; Butté, A.; et al. Process-wide control and automation of an integrated continuous manufacturing platform for antibodies. Biotechnol. Bioeng. 2020, 117, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Claßen, J.; Aupert, F.; Reardon, K.F.; Solle, D.; Scheper, T. Spectroscopic sensors for in-line bioprocess monitoring in research and pharmaceutical industrial application. Anal. Bioanal. Chem. 2017, 409, 651–666. [Google Scholar] [CrossRef]
- Kornecki, M.; Strube, J. Process Analytical Technology for Advanced Process Control in Biologics Manufacturing with the Aid of Macroscopic Kinetic Modeling. Bioengineering 2018, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.M.; Kessler, J.-M.; Salou, P.; Menezes, J.C.; Peinado, A. Monitoring mAb cultivations with in-situ raman spectroscopy: The influence of spectral selectivity on calibration models and industrial use as reliable PAT tool. Biotechnol. Prog. 2018, 34, 659–670. [Google Scholar] [CrossRef]
- Abu-Absi, N.R.; Kenty, B.M.; Cuellar, M.E.; Borys, M.C.; Sakhamuri, S.; Strachan, D.J.; Hausladen, M.C.; Li, Z.J. Real time monitoring of multiple parameters in mammalian cell culture bioreactors using an in-line Raman spectroscopy probe. Biotechnol. Bioeng. 2011, 108, 1215–1221. [Google Scholar] [CrossRef]
- Suvarov, P.; Wouwer, A.V.; Lee, J.W.; Seidel-Morgensten, A.; Kienle, A. Control of incomplete separation in simulated moving bed chromatographic processes. IFAC-PapersOnLine 2016, 49, 153–158. [Google Scholar] [CrossRef]
- Engell, S.; Toumi, A. Optimization and control of chromatography. In European Symposium on Computer-Aided Process Engineering—14, Proceedings of the 37th European Symposium of the Working Party on Computer-Aided Process Engineering, ESCAPE-14, Lisbon, Portugal, 16–19 May 2004, 1st ed.; Barbosa-Póvoa, A., Matos, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 9–22. ISBN 9780444516947. [Google Scholar]
- Strube, J.; Klatt, K.U.; Noeth, G.; Greifenberg, J.; Bocker, S.; Kansy, H.; Jahn, P.; Justen, B. Modular Valve System for Countercurrent Chromatography Process. U.S. Patent 8,658,040, 25 February 2014. [Google Scholar]
- Böcker, S.; Greifenberg, J.; Jähn, P.; Justen, B.; Kansy, H.; Klatt, K.; Noeth, G.; Strube, J. Process for the Preparation of Chemical and Pharmaceutical Products with Integrated Multi-Column Chromatography. Patent DE102004025000A1, 8 December 2005. [Google Scholar]
- Brestrich, N.; Briskot, T.; Osberghaus, A.; Hubbuch, J. A tool for selective inline quantification of co-eluting proteins in chromatography using spectral analysis and partial least squares regression. Biotechnol. Bioeng. 2014, 111, 1365–1373. [Google Scholar] [CrossRef]
- Buijs, J.; Norde, W.; Lichtenbelt, J.W.T. Changes in the Secondary Structure of Adsorbed IgG and F(ab‘)2 Studied by FTIR Spectroscopy. Langmuir 1996, 12, 1605–1613. [Google Scholar] [CrossRef]
- ICH. Endorsed Guide for ICH Q8/Q9/Q10 Implementation; International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use: Geneva, Switzerland , 2011. [Google Scholar]
- Wasalathanthri, D.P.; Feroz, H.; Puri, N.; Hung, J.; Lane, G.; Holstein, M.; Chemmalil, L.; Both, D.; Ghose, S.; Ding, J.; et al. Real-time monitoring of quality attributes by in-line Fourier transform infrared spectroscopic sensors at ultrafiltration and diafiltration of bioprocess. Biotechnol. Bioeng. 2020, 117, 3766–3774. [Google Scholar] [CrossRef]
- Sixt, M.; Uhlenbrock, L.; Strube, J. Toward a Distinct and Quantitative Validation Method for Predictive Process Modelling—On the Example of Solid-Liquid Extraction Processes of Complex Plant Extracts. Processes 2018, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Lohmann, L.J.; Strube, J. Accelerating Biologics Manufacturing by Modeling: Process Integration of Precipitation in mAb Downstream Processing. Processes 2020, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Klepzig, L.S.; Juckers, A.; Knerr, P.; Harms, F.; Strube, J. Digital Twin for Lyophilization by Process Modeling in Manufacturing of Biologics. Processes 2020, 8, 1325. [Google Scholar] [CrossRef]
- Uhlenbrock, L.; Jensch, C.; Tegtmeier, M.; Strube, J. Digital Twin for Extraction Process Design and Operation. Processes 2020, 8, 866. [Google Scholar] [CrossRef]
- Huter, M.J.; Jensch, C.; Strube, J. Model Validation and Process Design of Continuous Single Pass Tangential Flow Filtration Focusing on Continuous Bioprocessing for High Protein Concentrations. Processes 2019, 7, 781. [Google Scholar] [CrossRef] [Green Version]
- Helgers, H.; Schmidt, A.; Lohmann, L.J.; Vetter, F.L.; Juckers, A.; Jensch, C.; Mouellef, M.; Zobel-Roos, S.; Strube, J. Towards Autonomous Operation by Advanced Process Control—Process Analytical Technology for Continuous Biologics Antibody Manufacturing. Processes 2021, 9, 172. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
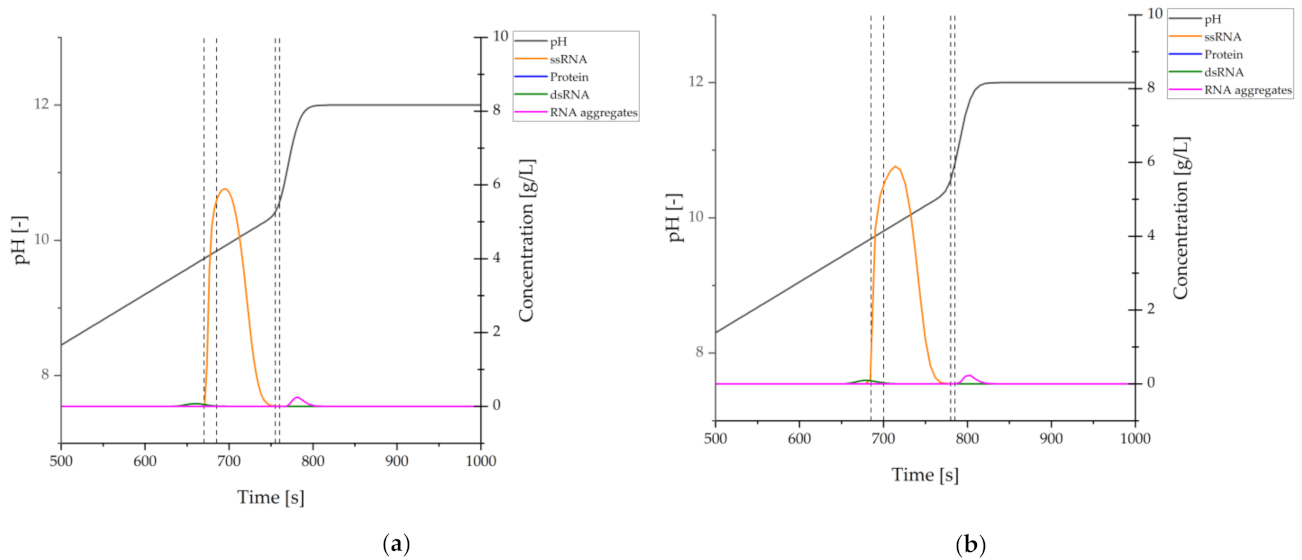{kind=link}
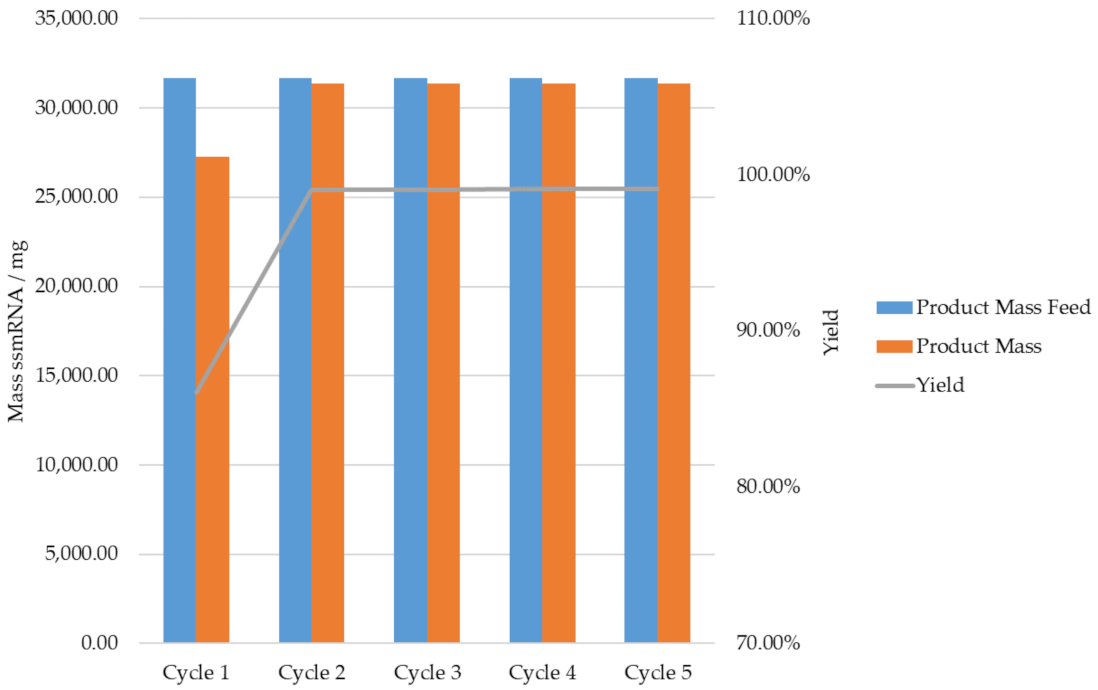{kind=link}
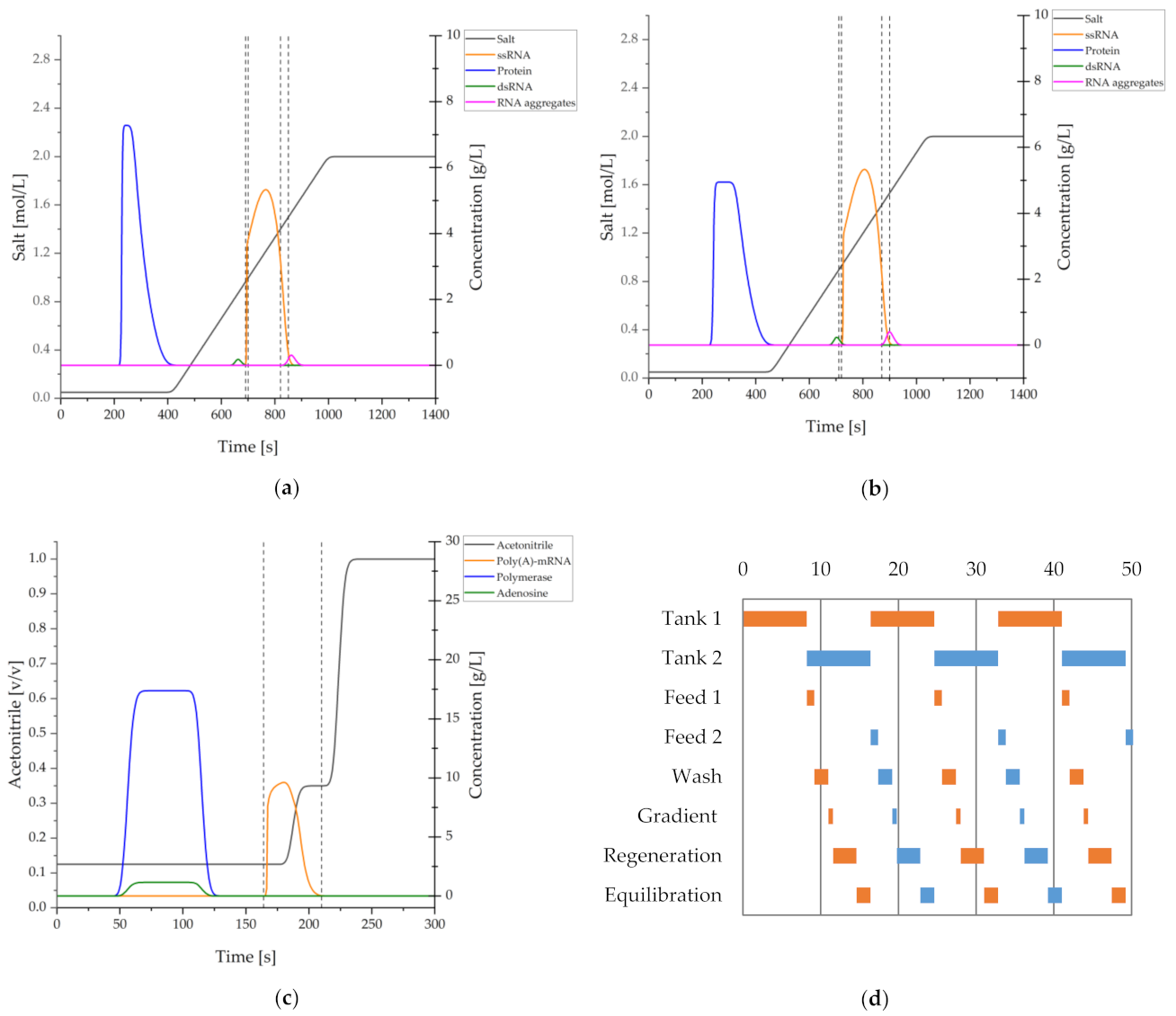{kind=link}
| Scenario | Batch | Mixed | Continuous |
|---|---|---|---|
| OPEX (M EUR/year) | 454.3 | 352.5 | 385.4 |
| CAPEX (M EUR) | 46.5 | 43.7 | 34.8 |
| Production Costs per dose (EUR) | 0.380 | 0.295 | 0.295 |
| Scenario | Processed mRNA | Input Downstream | Runs MMC | CV/h MMC | Flow Rate MMC | Input RP | Runs MMC | CV/h RP | Flow Rate RP |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 126.50 g/h | 16.67 L/h | 4.6 1/h | 122.8 1/h | 982.4 L/h | 39.27 L/h | 1.6 1/h | 39.4 1/h | 314.9 L/h |
| 2 | 75.90 g/h | 10.00 L/h | 2.8 1/h | 73.7 1/h | 589.5 L/h | 23.56 L/h | 1.0 1/h | 23.6 1/h | 188.9 L/h |
| 3 | 63.25 g/h | 8.33 L/h | 2.3 1/h | 61.4 1/h | 491.2 L/h | 19.64 L/h | 0.8 1/h | 19.7 1/h | 157.4 L/h |
| 4 | 58.38 g/h | 7.69 L/h | 2.1 1/h | 56.7 1/h | 453.4 L/h | 18.13 L/h | 0.8 1/h | 18.2 1/h | 145.3 L/h |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, A.; Helgers, H.; Vetter, F.L.; Juckers, A.; Strube, J. Fast and Flexible mRNA Vaccine Manufacturing as a Solution to Pandemic Situations by Adopting Chemical Engineering Good Practice—Continuous Autonomous Operation in Stainless Steel Equipment Concepts. Processes 2021, 9, 1874. https://doi.org/10.3390/pr9111874
Schmidt A, Helgers H, Vetter FL, Juckers A, Strube J. Fast and Flexible mRNA Vaccine Manufacturing as a Solution to Pandemic Situations by Adopting Chemical Engineering Good Practice—Continuous Autonomous Operation in Stainless Steel Equipment Concepts. Processes. 2021; 9(11):1874. https://doi.org/10.3390/pr9111874
Chicago/Turabian StyleSchmidt, Axel, Heribert Helgers, Florian Lukas Vetter, Alex Juckers, and Jochen Strube. 2021. "Fast and Flexible mRNA Vaccine Manufacturing as a Solution to Pandemic Situations by Adopting Chemical Engineering Good Practice—Continuous Autonomous Operation in Stainless Steel Equipment Concepts" Processes 9, no. 11: 1874. https://doi.org/10.3390/pr9111874