


Mathematical Modeling of Eicosanoid Metabolism in Macrophage Cells: Cybernetic Framework Combined with Novel Information-Theoretic Approaches

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Development of the Cybernetic Formulation: Kinetic Model
2.1.1. Kinetic Equations for PRs Branch
2.1.2. Enzyme Balance
2.1.3. Cybernetic Goal Formulation and Cybernetic Control Variables and
2.2. Development of Information-Theoretic Approaches for Formulating the Cybernetic Goal
2.2.1. Data Needed for the Information-Theoretic Approach
2.2.2. Rényi Symbolic Transfer Entropy (RSTE): Derivation of Causality
2.2.3. Branch-Specific Cybernetic Goals for PRs and LTs Branches
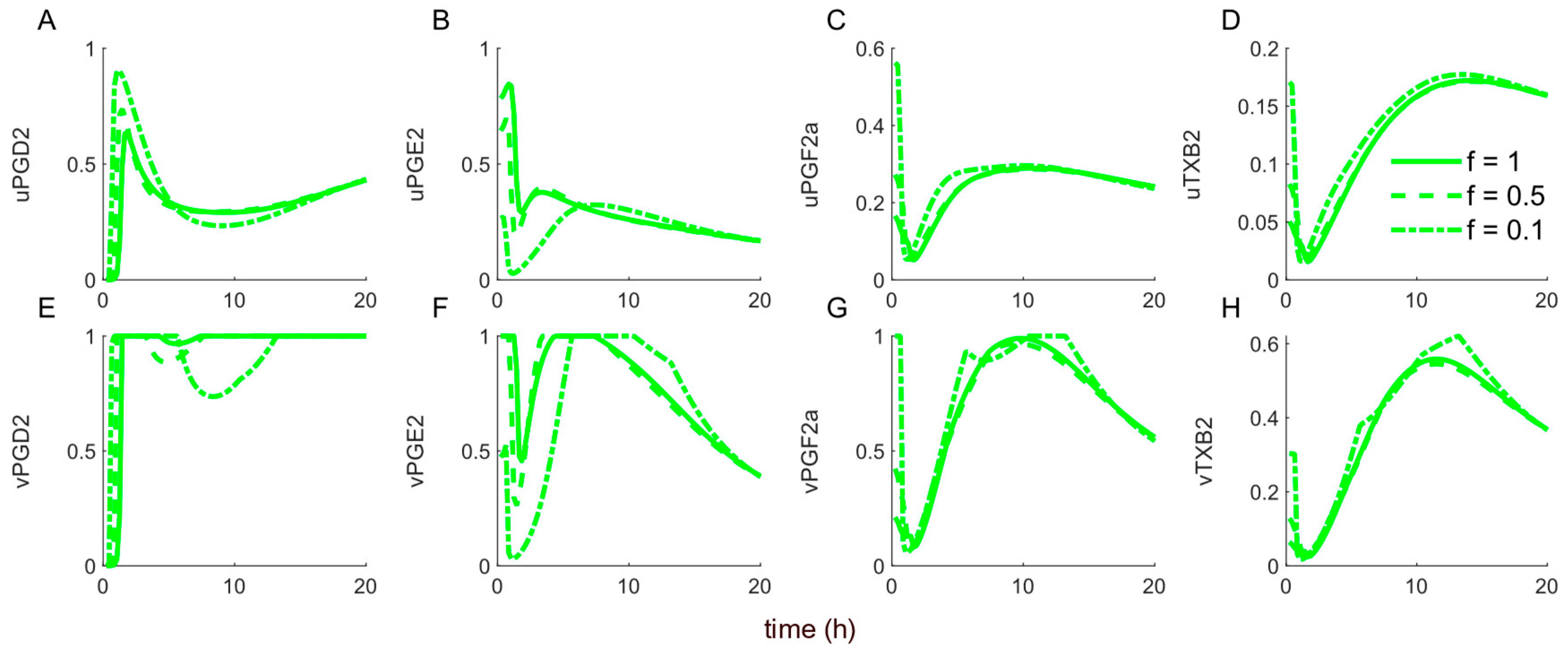
2.2.4. Weight Calculation Using Mutual Information
2.3. Simulation Strategy for the Cybernetic Framework (Figure 2)

2.4. Model Validation
2.4.1. Perturbation Experiment: Gene Knockout (Chemical Knockdown in Supplementary, Section S4)
3. Results
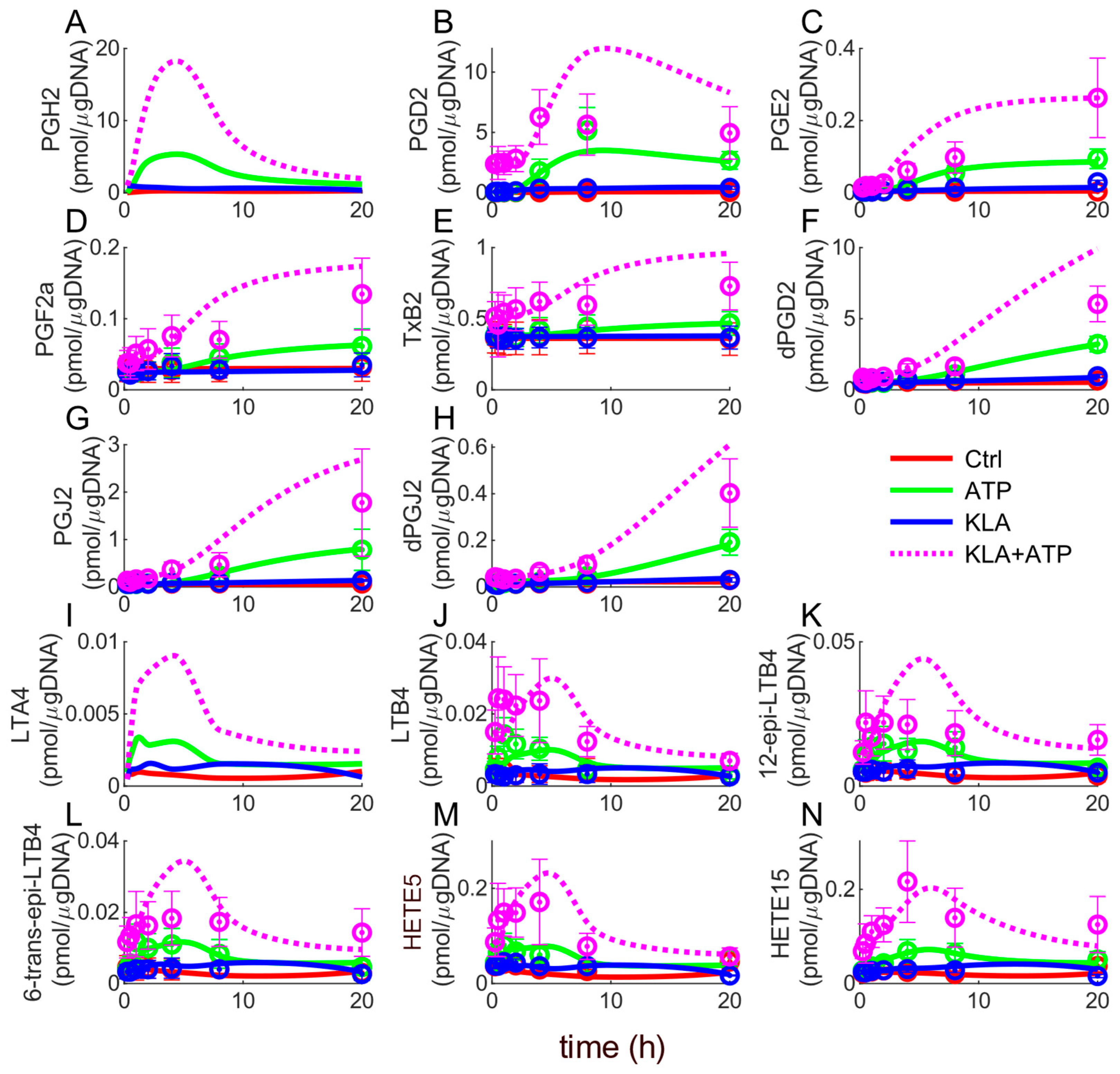
3.1. Simulated Profiles and Model Validation
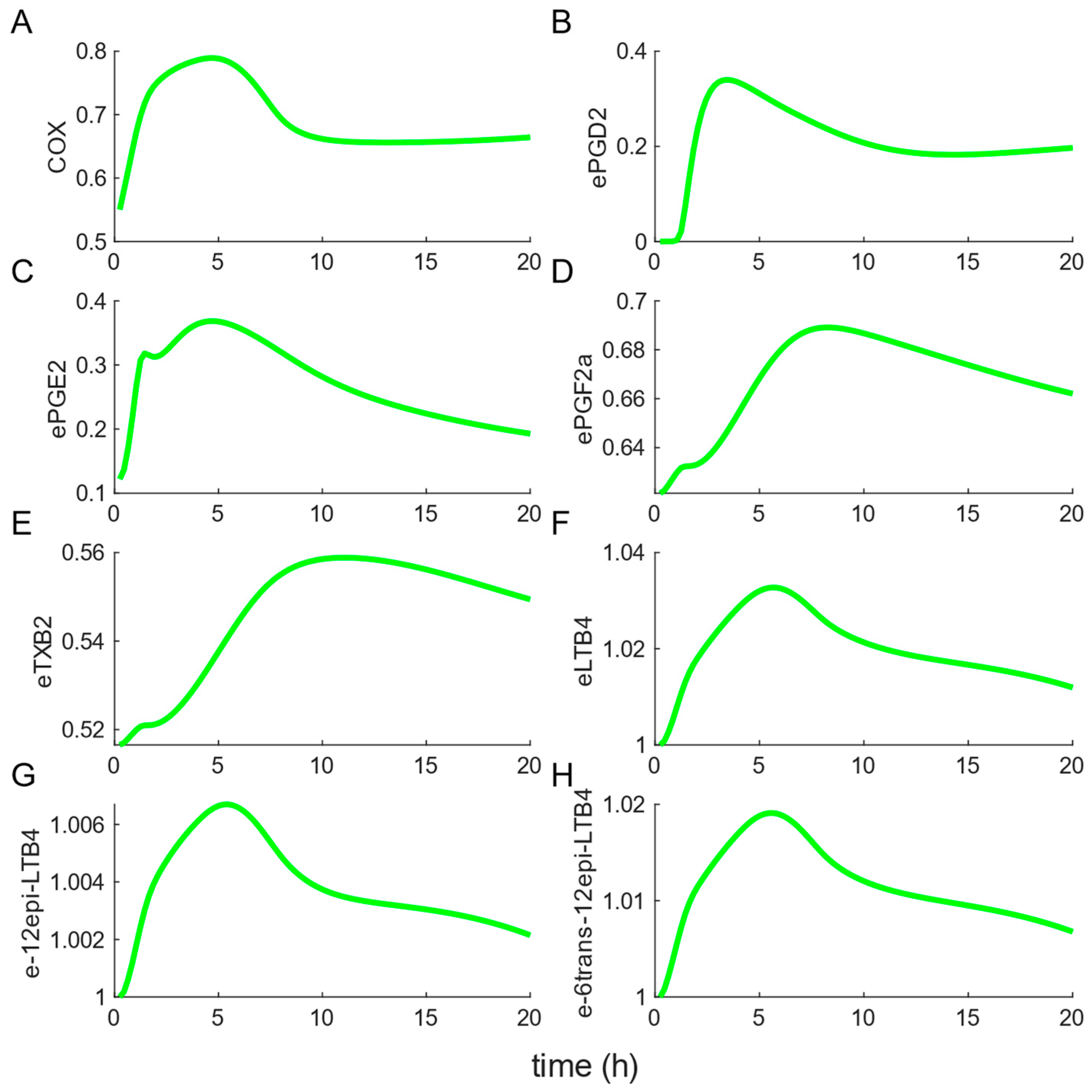
3.2. Model Outcome: Prediction of Enzyme Dynamics
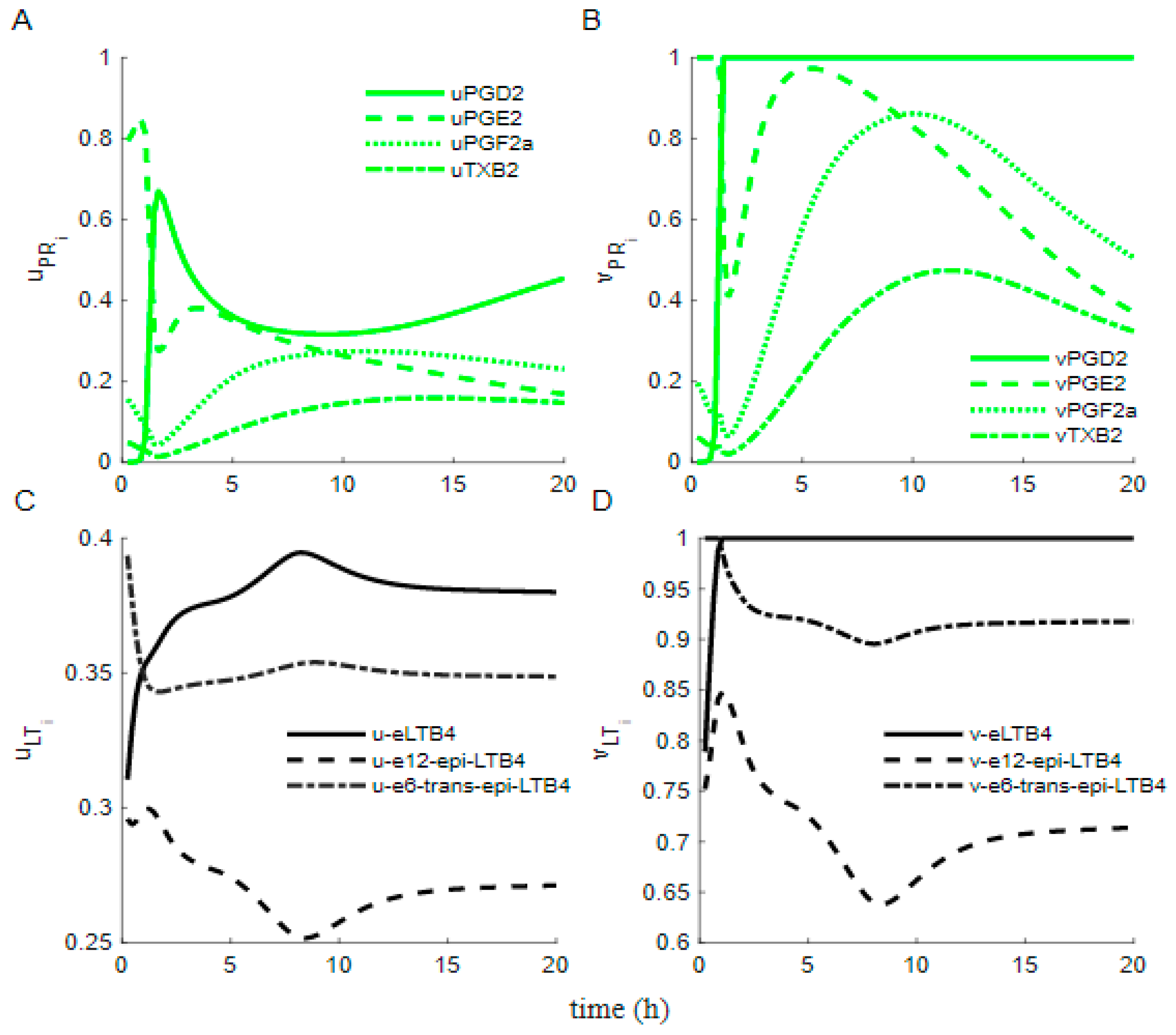
3.3. Dynamics of the Cybernetic Control Variables ,
4. Model Validation: Model Perturbation Experiments
4.1. Gene Knockdown: Enzyme Synthesis Perturbation of Ltbd4h (eLTB4) & Ptges (ePGE2)
4.1.1. Ltbd4h (eLTB4) Inhibition
4.1.2. Results of Ptges (ePGE2) Inhibition
4.2. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dennis, E.; Norris, P. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Hammock, B.D.; Wang, W.; Gilligan, M.M.; Panigrahy, D. Eicosanoids: The Overlooked Storm in Coronavirus Disease 2019 (COVID-19)? Am. J. Pathol. 2020, 190, 1782–1788. [Google Scholar] [CrossRef]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality, Inflamm. Regen 2020, 40, 37. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Morgulchik, N.; Athanasopoulou, F.; Chu, E.; Lam, Y.; Kamaly, N. Potential therapeutic approaches for targeted inhibition of inflammatory cytokines following COVID-19 infection-induced cytokine storm. Interface Focus 2021, 12, 20210006. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Wang, G.Y.; Zhao, R.; An, Z.L.; Liu, L.H. Eicosanoid Metabolomic Profile of Remdesivir Treatment in Rat Plasma by High-Performance Liquid Chromatography Mass Spectrometry. Front. Pharmacol. 2021, 12, 747450. [Google Scholar] [CrossRef]
- Yiu, H.H.; Graham, A.L.; Stengel, R.F. Dynamics of a Cytokine Storm. PLoS ONE 2012, 7, e45027. [Google Scholar] [CrossRef]
- Blaho, V.A.; Buczynski, M.W.; Brown, C.R.; Dennis, E.A. Lipidomic analysis of dynamic eicosanoid responses during the induction and resolution of lyme arthritis. J. Biol. Chem. 2009, 284, 21599–21612. [Google Scholar] [CrossRef] [Green Version]
- Young, J.D.; Ramkrishna, D. On the Matching and Proportional Laws of Cybernetic Models. Biotechnol. Prog. 2007, 23, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Song, D.R.H.-S. Dynamic Models of Metabolism: Review of the Cybernetic Approach Doraiswami. AIChE J. 2012, 59, 215–228. [Google Scholar] [CrossRef]
- Ramkrishna, D.; Song, H.-S. Cybernetic Modeling for Bioreaction Engineering; Cambridge University Press: New York, NY, USA, 2018. [Google Scholar] [CrossRef] [Green Version]
- Aboulmouna, L.; Raja, R.; Khanum, S.; Gupta, S.; Maurya, M.R.; Grama, A.; Subramaniam, S.; Ramkrishna, D. Cybernetic modeling of biological processes in mammalian systems. Curr. Opin. Chem. Eng. 2020, 30, 120–127. [Google Scholar] [CrossRef]
- Baral, S.; Raja, R.; Sen, P.; Dixit, N.M. Towards multiscale modeling of the CD8+ T cell response to viral infections, Wiley Interdiscip. Rev. Syst. Biol. Med. 2019, 11, e1446. [Google Scholar] [CrossRef] [Green Version]
- Yizhak, K.; Chaneton, B.; Gottlieb, E.; Ruppin, E. Modeling cancer metabolism on a genome scale. Mol. Syst. Biol. 2015, 11, 817. [Google Scholar] [CrossRef]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nature Biotechnology: Nature Publishing Group. Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Raman, K.; Chandra, N. Flux balance analysis of biological systems: Applications and challenges. Brief. Bioinform. 2009, 10, 435–449. [Google Scholar] [CrossRef]
- Bornheimer, S.J.; Maurya, M.R.; Farquhar, M.G.; Subramaniam, S. Computational modeling reveals how interplay between components of a GTPase-cycle module regulates signal transduction. Proc. Natl. Acad. Sci. USA 2004, 101, 15899–15904. [Google Scholar] [CrossRef] [Green Version]
- Maurya, M.R.; Subramaniam, S. A kinetic model for calcium dynamics in RAW 264.7 cells: 1. Mechanisms, parameters, and subpopulational variability. Biophys. J. 2007, 93, 709–728. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Maurya, M.R.; Stephens, D.L.; Dennis, E.A.; Subramaniam, S. An integrated model of eicosanoid metabolism and signaling based on lipidomics flux analysis. Biophys. J. 2009, 96, 4542–4551. [Google Scholar] [CrossRef] [Green Version]
- Hanly, T.J.; Henson, M.A. Dynamic flux balance modeling of microbial co-cultures for efficient batch fermentation of glucose and xylose mixtures. Biotechnol. Bioeng. 2011, 108, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, S.; Price, N.D. Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 17845–17850. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.; Fell, D.A.; Dandekar, T. A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nat. Biotechnol. 2000, 18, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Aboulmouna, L.; Gupta, S.; Maurya, M.R.; DeVilbiss, F.T.; Subramaniam, S.; Ramkrishna, D. A cybernetic approach to modeling lipid metabolism in mammalian cells. Processes 2018, 6, 126. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishna, R.; Ramakrishna, D.; Konopka, A.E. Cybernetic modeling of growth in mixed, substitutable substrate environments: Preferential and simultaneous utilization. Biotechnol. Bioeng. 1996, 52, 141–151. [Google Scholar] [CrossRef]
- Varner, J.D. Large-scale prediction of phenotype: Concept. Biotechnol. Bioeng. 2000, 69, 664–678. [Google Scholar] [CrossRef]
- Varner, J.; Ramkrishna, D. Metabolic Engineering from a Cybernetic Perspective: Aspartate Family of Amino Acids. Metab. Eng. 1999, 1, 88–116. [Google Scholar] [CrossRef] [Green Version]
- Song, H.S.; Ramkrishna, D. Prediction of dynamic behavior of mutant strains from limited wild-type data. Metab. Eng. 2012, 14, 69–80. [Google Scholar] [CrossRef]
- Song, H.S.; Ramkrishna, D. Cybernetic models based on lumped elementary modes accurately predict strain-specific metabolic function. Biotechnol. Bioeng. 2011, 108, 127–140. [Google Scholar] [CrossRef]
- Snodgrass, R.G.; Brüne, B. Regulation and Functions of 15-Lipoxygenases in Human Macrophages. Front. Pharmacol. 2019, 10, 719. [Google Scholar] [CrossRef] [Green Version]
- Hanna, V.S.; Hafez, E.A.A. Synopsis of arachidonic acid metabolism: A review. J. Adv. Res. 2018, 11, 23–32. [Google Scholar] [CrossRef]
- Bennett, M.; Gilroy, D.W. Lipid Mediators in Inflammation. ASM J. 2016, 4, 1–21. [Google Scholar] [CrossRef]
- Werner, M.; Jordan, P.M.; Romp, E.; Czapka, A.; Rao, Z.; Kretzer, C.; Koeberle, A.; Garscha, U.; Pace, S.; Claesson, H.E.; et al. Targeting biosynthetic networks of the proinflammatory and proresolving lipid metabolome. FASEB J. 2019, 33, 6140–6153. [Google Scholar] [CrossRef]
- Meng, H.; Liu, Y.; Lai, L. Diverse Ways of Perturbing the Human Arachidonic Acid Metabolic Network to Control Inflammation. Acc. Chem. Res. 2015, 48, 2242–2250. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Fu, X.; Chen, Q.; Patra, J.K.; Wang, D.; Wang, Z.; Gai, Z. Arachidonic acid metabolism and kidney inflammation. Int. J. Mol. Sci. 2019, 20, 3683. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, V.B.; Rossjohn, J.; Wakelam, M.J.O. Phospholipid signaling in innate immune cells. J. Clin. Investig. 2018, 128, 2670–2679. [Google Scholar] [CrossRef] [PubMed]
- Sala, A.; Proschak, E.; Steinhilber, D.; Rovati, G.E. Two-pronged approach to anti-inflammatory therapy through the modulation of the arachidonic acid cascade. Biochem. Pharmacol. 2018, 158, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kihara, Y.; Gupta, S.; Maurya, M.R.; Armando, A.; Shah, I.; Quehenberger, O.; Glass, C.K.; Dennis, E.A.; Subramaniam, S. Modeling of eicosanoid fluxes reveals functional coupling between cyclooxygenases and terminal synthases. Biophys. J. 2014, 106, 966–975. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kihara, Y.; Maurya, M.R.; Norris, P.C.; Dennis, E.A.; Subramaniam, S. Computational Modeling of Competitive Metabolism between ω3- and ω6-Polyunsaturated Fatty Acids in Inflammatory Macrophages. J. Phys. Chem. B 2016, 120, 8346–8353. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Wang, X.; Li, K.; Liu, Y.; Liu, Y.; Wang, R.; Li, H. Pharmacodynamics simulation of HOEC by a computational model of arachidonic acid metabolic network. Quant. Biol. 2019, 7, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Ma, W.; Liang, H.; Ouyang, Q.; Tang, C.; Lai, L. Dynamic simulations on the arachidonic acid metabolic network. PLoS Comput. Biol. 2007, 3, 0523–0530. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Hide, I.; Ido, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Production and Release of Neuroprotective Tumor Necrosis Factor by P2X 7 Receptor-Activated Microglia. J. Neurosci. 2004, 24, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Bruggen, T.; Nijenhuis, S.; Van, E.; Verhoef, J.; Van Asbeck, B.S. Lipopolysaccharide-Induced Tumor Necrosis Factor Alpha Production by Human Monocytes Involves the Raf-1/MEK1-MEK2/ERK1-ERK2 Pathway. Infect. Immun. 1999, 67, 3824–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, K.; Hwang, J.J.; Kwon, B.I.; Kheradmand, F.; Corry, D.B.; Lee, S.H. Leukotriene enhanced allergic lung inflammation through induction of chemokine production. Clin. Exp. Med. 2015, 15, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.H.; Shimizu, T.; Spener, F.; Van Meer, G.; Wakelam, M.J.O.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, 527–532. [Google Scholar] [CrossRef] [Green Version]
- Sud, M.; Fahy, E.; Cotter, D.; Dennis, E.A.; Subramaniam, S. LIPID MAPS-nature lipidomics gateway: An online resource for students and educators interested in lipids. J. Chem. Educ. 2012, 89, 291–292. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Maurya, M.R.; Merrill, A.H.; Glass, C.K.; Subramaniam, S.; Merrill, A.H., Jr.; Glass, C.K.; Subramaniam, S. Integration of lipidomics and transcriptomics data towards a systems biology model of sphingolipid metabolism. BMC Syst. Biol. 2011, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekharan, N.V.; Dai, H.; Roos, K.L.T.; Evanson, N.K.; Tomsik, J.; Elton, T.S.; Simmons, D.L. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: Cloning, structure, and expression. Proc. Natl. Acad. Sci. USA 2002, 99, 13926–13931. [Google Scholar] [CrossRef] [Green Version]
- Straight, J.V.; Ramkrishna, D. Cybernetic Modeling and Regulation of Metabolic Pathways. Growth on Complementary Nutrients. Biotechnol. Prog. 1994, 10, 574–587. [Google Scholar] [CrossRef]
- Papana, A.; Kyrtsou, C.; Kugiumtzis, D.; Diks, C. Detecting Causality in Non-stationary Time Series Using Partial Symbolic Transfer Entropy: Evidence in Financial Data. Comput. Econ. 2016, 47, 341–365. [Google Scholar] [CrossRef]
- Ursino, M.; Ricci, G.; Magosso, E. Transfer Entropy as a Measure of Brain Connectivity: A Critical Analysis with the Help of Neural Mass Models. Front. Comput. Neurosci. 2020, 14, 45. [Google Scholar] [CrossRef]
- McBride, J.; Zhao, X.; Munro, N.; Jicha, G.; Smith, C.; Jiang, Y. Discrimination of mild cognitive impairment and alzheimer’s disease using transfer entropy measures of scalp EEG. J. Healthc. Eng. 2015, 6, 55–70. [Google Scholar] [CrossRef]
- Zhu, R.; Hochstetter, J.; Loeffler, A.; Diaz-Alvarez, A.; Nakayama, T.; Lizier, J.T.; Kuncic, Z. Information dynamics in neuromorphic nanowire networks. Sci. Rep. 2021, 11, 13047. [Google Scholar] [CrossRef] [PubMed]
- Marschinski, R.; Kantz, H. Analysing the information flow between financial time series. Eur. Phys. J. B 2002, 30, 275–281. [Google Scholar] [CrossRef]
- Jizba, P.; Lavička, H.; Tabachová, Z. Rényi Transfer Entropy Estimators for Financial Time Series. In Proceedings of the 7th International Conference on Time Series and Forecasting, Gran Canaria, Spain, 19–21 July 2021; Volume 33. [Google Scholar] [CrossRef]
- Jizba, P.; Kleinert, H.; Shefaat, M. Rnyi’s information transfer between financial time series. Phys. A Stat. Mech. Its Appl. 2012, 391, 2971–2989. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Shang, P.; Lin, A. Financial time series analysis based on effective phase transfer entropy. Phys. A Stat. Mech. Its Appl. 2017, 468, 398–408. [Google Scholar] [CrossRef]
- Teng, Y.; Shang, P. Transfer entropy coefficient: Quantifying level of information flow between financial time series. Phys. A Stat. Mech. Its Appl. 2017, 469, 60–70. [Google Scholar] [CrossRef]
- Mao, X.; Shang, P. Transfer entropy between multivariate time series, Commun. Nonlinear Sci. Numer. Simul. 2017, 47, 338–347. [Google Scholar] [CrossRef]
- Kim, J.; Jakobsen, S.T.; Natarajan, K.N.; Won, K.J. TENET: Gene network reconstruction using transfer entropy reveals key regulatory factors from single cell transcriptomic data. Nucleic Acids Res. 2021, 49, e1. [Google Scholar] [CrossRef]
- Castro, J.C.; Valdés, I.; Gonzalez-García, L.N.; Danies, G.; Cañas, S.; Winck, F.V.; Ñústez, C.E.; Restrepo, S.; Riaño-Pachón, D.M. Gene regulatory networks on transfer entropy (GRNTE): A novel approach to reconstruct gene regulatory interactions applied to a case study for the plant pathogen Phytophthora infestans. Theor. Biol. Med. Model. 2019, 16, 7. [Google Scholar] [CrossRef] [Green Version]
- Staniek, M.; Lehnertz, K. Symbolic transfer entropy. Phys. Rev. Lett. 2008, 100, 158101. [Google Scholar] [CrossRef] [PubMed]
- Bandt, C.; Pompe, B. Permutation Entropy: A Natural Complexity Measure for Time Series. Phys. Rev. Lett. 2002, 88, 4. [Google Scholar] [CrossRef] [PubMed]
- Kraskov, A.; Stögbauer, H.; Grassberger, P. Estimating mutual information, Phys. Rev. E-Stat. Physics, Plasmas, Fluids. Relat. Interdiscip. Top. 2004, 69, 16. [Google Scholar] [CrossRef] [Green Version]
- Kvålseth, T. On Normalized Mutual Information: Measure Derivations and Properties. Entropy 2017, 19, 631. [Google Scholar] [CrossRef] [Green Version]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Technol. J. 1948, 27, 623–656. [Google Scholar] [CrossRef]
- Reshef, D.; Reshef, Y.; Finucane, H.; Grossman, S.; Mcvean, G.; Turnbaugh, P.; Lander, E.; Mitzenmacher, M.; Sabeti, P. Detecting Novel Associations in Large Data Sets. Sci. Transl. Med. 2011, 334, 1518–1524. [Google Scholar] [CrossRef] [Green Version]
- de Siqueira Santos, S.; Takahashi, D.; Nakata, A.; Fujita, A. A comparative study of statistical methods used to identify dependencies between gene expression signals. Brief. Bioinform. 2013, 15, 906–918. [Google Scholar] [CrossRef] [Green Version]
- Numata, J.; Ebenhöh, O.; Knapp, E.-W. Measuring Correlations in Metabolic Networks with Mutual Information. Angew. Chemie Int. Ed. 2008, 20, 112–122. [Google Scholar]
- Batina, L.; Gierlichs, B.; Prouff, E.; Rivain, M.; Standaert, F.X.; Veyrat-Charvillon, N. Mutual information analysis: A comprehensive study. J. Cryptol. 2011, 24, 269–291. [Google Scholar] [CrossRef] [Green Version]
- Galka, A.; Ozaki, T.; Bayard, J.B.; Yamashita, O. Whitening as a tool for estimating mutual information in spatiotemporal data sets. J. Stat. Phys. 2006, 124, 1275–1315. [Google Scholar] [CrossRef]
- Kigozi, R.; Zinszer, K.; Mpimbaza, A.; Sserwanga, A.; Kigozi, S.; Kamya, M. Assessing temporal associations between environmental factors and malaria morbidity at varying transmission settings in Uganda. Malar. J. 2016, 15, 4–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, W.; Brockwell, P.J.; Davis, R.A. Time Series: Theory and Methods; Springer Press: New York, NY, USA, 1989. [Google Scholar] [CrossRef]
- He, C.; Wu, Y.; Lai, Y.; Cai, Z.; Liu, Y.; Lai, L. Dynamic eicosanoid responses upon different inhibitor and combination treatments on the arachidonic acid metabolic network. Mol. Biosyst. 2012, 8, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Mazaleuskaya, L.L.; Lawson, J.A.; Li, X.; Grant, G.; Mesaros, C.; Grosser, T.; Blair, I.A.; Ricciotti, E.; FitzGerald, G.A. A broad-spectrum lipidomics screen of antiinflammatory drug combinations in human blood. JCI Insight. 2016, 1, e87031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Bai, H.; Ouyang, Q.; Lai, L.; Tang, C. Finding multiple target optimal intervention in disease-related molecular network. Mol. Syst. Biol. 2008, 4, 228. [Google Scholar] [CrossRef]
- CBuckley, D.; Gilroy, D.W.; Serhan, C.N. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aboulmouna, L.; Khanum, S.; Heidari, M.; Raja, R.; Gupta, S.; Maurya, M.R.; Grama, A.; Subramaniam, S.; Ramkrishna, D. Mathematical Modeling of Eicosanoid Metabolism in Macrophage Cells: Cybernetic Framework Combined with Novel Information-Theoretic Approaches. Processes 2023, 11, 874. https://doi.org/10.3390/pr11030874
Aboulmouna L, Khanum S, Heidari M, Raja R, Gupta S, Maurya MR, Grama A, Subramaniam S, Ramkrishna D. Mathematical Modeling of Eicosanoid Metabolism in Macrophage Cells: Cybernetic Framework Combined with Novel Information-Theoretic Approaches. Processes. 2023; 11(3):874. https://doi.org/10.3390/pr11030874
Chicago/Turabian StyleAboulmouna, Lina, Sana Khanum, Mohsen Heidari, Rubesh Raja, Shakti Gupta, Mano R. Maurya, Ananth Grama, Shankar Subramaniam, and Doraiswami Ramkrishna. 2023. "Mathematical Modeling of Eicosanoid Metabolism in Macrophage Cells: Cybernetic Framework Combined with Novel Information-Theoretic Approaches" Processes 11, no. 3: 874. https://doi.org/10.3390/pr11030874