
The Perspective of Using the System Ethanol-Ethyl Acetate in a Liquid Organic Hydrogen Carrier (LOHC) Cycle
 ,
,  , and
, and
Abstract
:1. Introduction
2. Methodology of Ethanol Dehydrogenation
2.1. Thermodynamics of the Occurring Reactions
2.2. Catalysts Normally Employed for Promoting the Ethanol Dehydrogenation
2.3. A Reference Catalyst, Reaction Mechanism and Related Kinetic Model
2.4. Laboratory and Pilot Chemical Plants
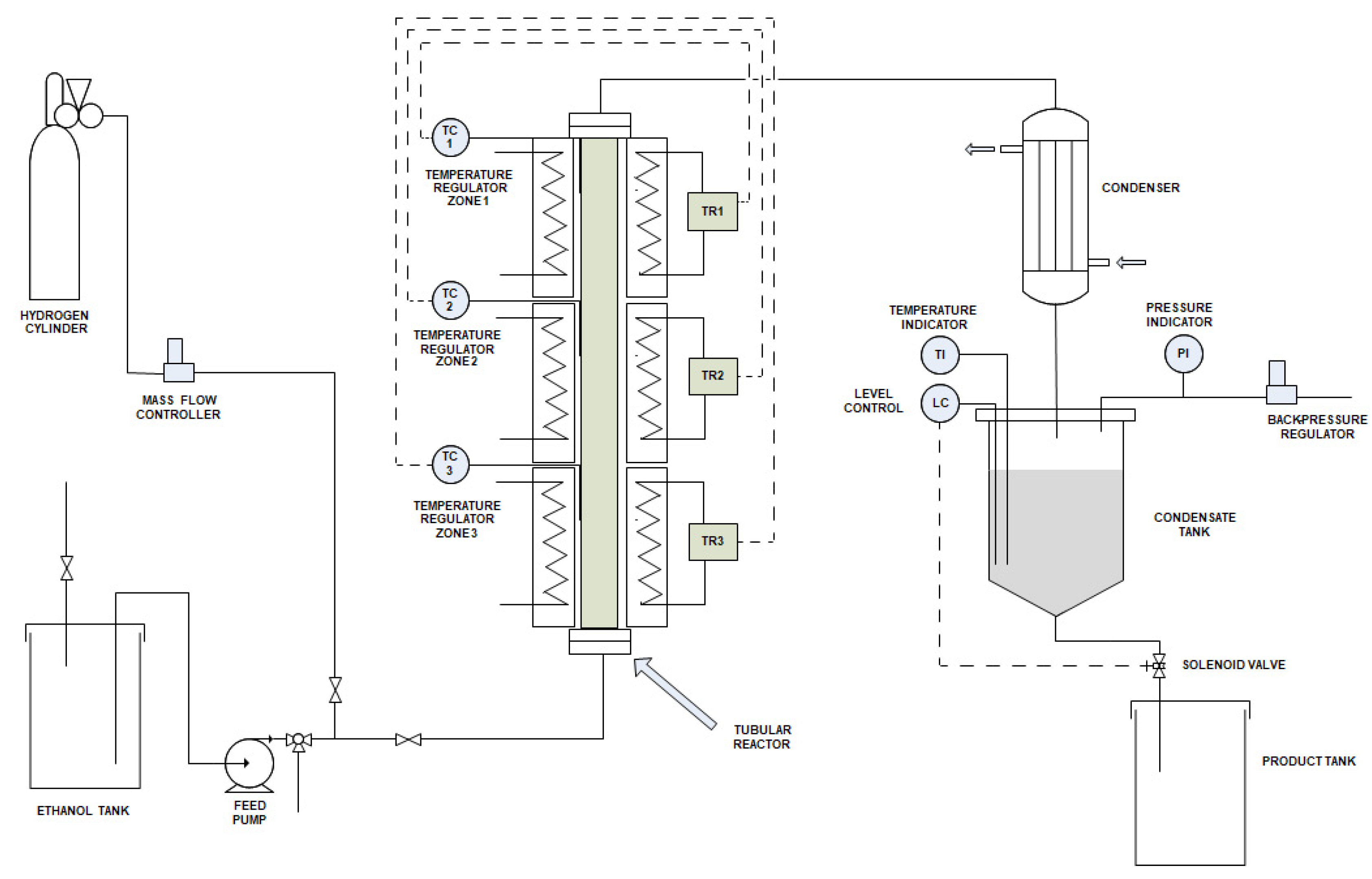
2.5. Scale-Up: Scheme of the Industrial Plant and Most Opportune Operative Conditions
2.6. Techno-Economic Analysis of the Process
- Only ethanol is required as feedstock. Then, if ethanol derives from a biological source the process is based on a renewable resource.
- No problems of corrosion are envisaged during the plant operation. This allows using lower grades of material in the equipment fabrication and the plant costs are therefore reduced.
- Pure hydrogen is obtained exempt from CO and CO2 and is therefore directly usable in fuel cells.
- High purity of the product ethyl acetate (>99%), which can be directly sent to the second hydrogen reloading step without significant action.
2.7. Process Intensification and Adoption of Membrane Reactors
3. Ethyl Acetate Hydrogenation
3.1. Thermodynamics of the Occurring Reactions
3.2. Catalysts Normally Employed for Promoting the Ethyl Acetate Hydrogenation Reaction
3.3. Ethyl Acetate Hydrogenation, Kinetic Methodology in Conventional Reactors
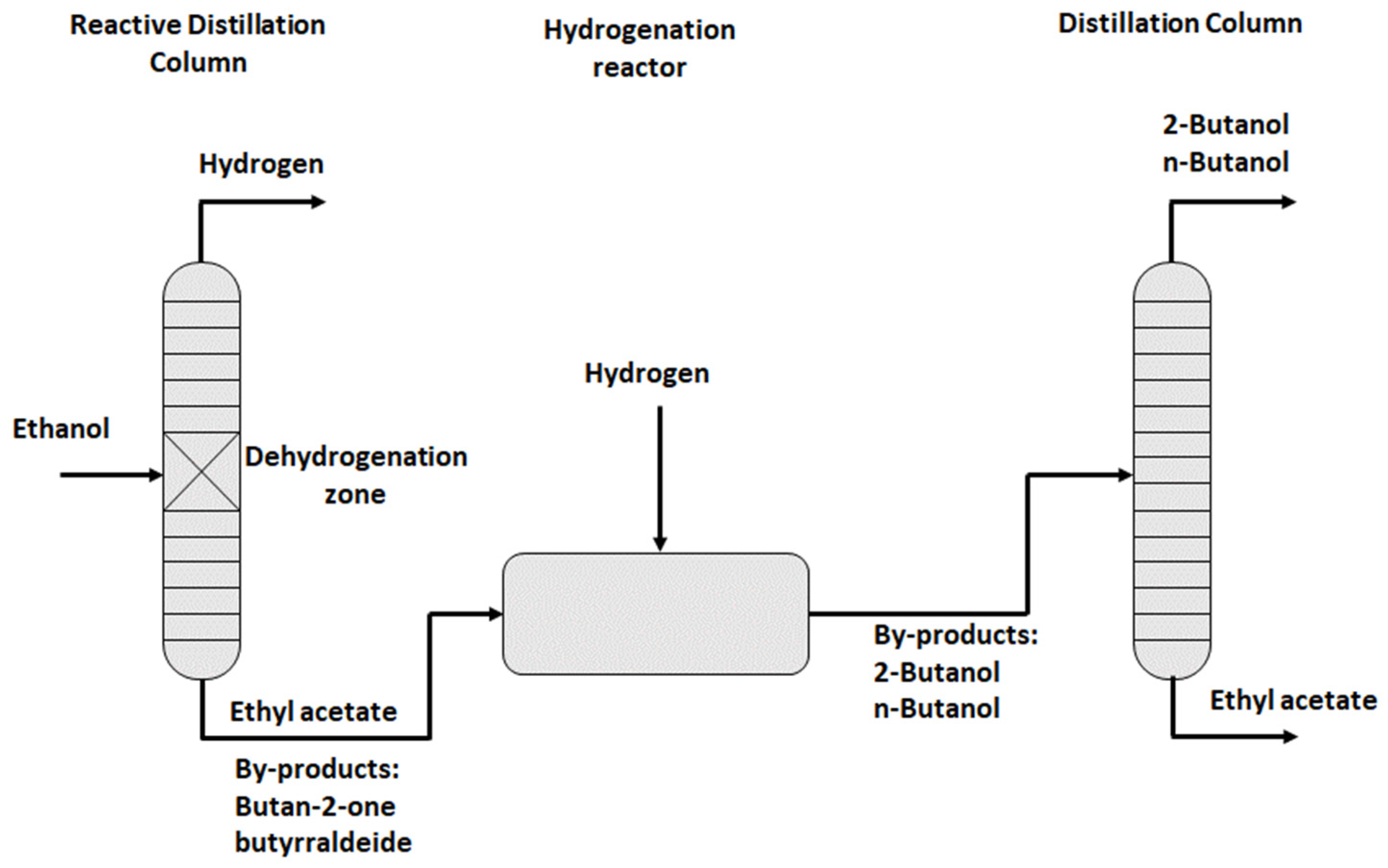
3.4. The Industrial Ethanol-Ethylacetate LOHC Process
4. Ethanol as a Possible LOHC in Comparison with Other Candidate Molecules
5. Conclusions
- (a)
- The realization of an innovative and simple system for hydrogen storage and transportation.
- (b)
- The use of inexpensive raw materials (bio-ethanol, ethyl acetate), which are abundant, renewable, biodegradable, non-toxic, and not dangerous for the environment.
- (c)
- The synthesis of more active and selective dehydrogenation-hydrogenation catalysts and the development of techniques for their characterization and kinetic testing.
- (d)
- The realization and modeling of membrane reactors for process intensification.
- (e)
- Modeling of the overall cyclic system for different operative scales.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- OECD/Food and Agriculture Organization of the United Nations. Biofuels. In OECD-FAO Agricultural Outlook 2022–2031; OECD Publishing: Paris, France, 2022; pp. 232–243. [Google Scholar] [CrossRef]
- Iwasa, N.; Takezawa, N. Reforming of ethanol—Dehydrogenation to ethyl acetate and steam reforming to acetic acid over copper-based catalysts. Bull. Chem. Soc. Jpn. 1991, 64, 2619–2623. [Google Scholar] [CrossRef]
- Tu, Y.I.; Chen, Y.W.; Li, C. Characterization of unsupported copper-chromium catalysts for ethanol dehydrogenation. J. Mol. Catal. 1994, 89, 179–189. [Google Scholar] [CrossRef]
- Wu, R.; Sun, K.; Chen, Y.; Zhang, M.; Wang, L. Ethanol dimerization to Ethyl acetate and hydrogen on the multifaceted copper catalysts. Surface Sci. 2021, 703, 121742. [Google Scholar] [CrossRef]
- Inui, K.; Kurabayashi, T.; Sato, S. Direct synthesis of ethyl acetate from ethanol over Cu-Zn-Zr-Al-O catalyst. Appl. Catal. A Gen. 2002, 237, 53–61. [Google Scholar] [CrossRef]
- Inui, K.; Kurabayashi, T.; Sato, S. Direct synthesis of ethyl acetate from ethanol carried out under pressure. J. Catal. 2002, 212, 207–215. [Google Scholar] [CrossRef]
- Inui, K.; Kurabayashi, T.; Sato, S.; Ichikawa, N. Effective formation of ethyl acetate from ethanol over Cu-Zn-Zr-Al-O catalyst. J. Mol. Catal. A Chem. 2004, 216, 147–156. [Google Scholar] [CrossRef]
- Colley, S.W.; Tabatabaei, J.; Waugh, K.C.; Wood, M.A. The detailed kinetics and mechanism of ethyl ethanoate synthesis over a Cu/Cr2O3 catalyst. J. Catal. 2005, 236, 21–33. [Google Scholar] [CrossRef]
- Gaspar, A.B.; Barbosa, F.G.; Letichevsky, S.; Appel, L.G. The one-pot ethyl acetate syntheses: The role of the support in the oxidative and the dehydrogenative routes. Appl. Catal. A Gen. 2010, 380, 113–117. [Google Scholar] [CrossRef]
- Semenov, I.P.; Men’shchikov, V.A.; Sycheva, O.I. Pilot tests of a catalyst for the production of ethyl acetate from ethanol. Catal. Ind. 2020, 12, 287–291. [Google Scholar] [CrossRef]
- Usman, M.R. Hydrogen storage methods: Review and current status. Renew. Sustain. Energy Rev. 2022, 167, 112743. [Google Scholar] [CrossRef]
- Phung, T.K. Copper-based catalysts for ethanol dehydrogenation and dehydrogenative coupling into hydrogen, acetaldehyde and ethyl acetate. Int. J. Hydrogen Energy 2022, 47, 42234–42249. [Google Scholar] [CrossRef]
- Sekine, Y.; Higo, T. Recent trends on the dehydrogenation catalysis of liquid organic hydrogen carrier (LOHC): A review. Top. Catal. 2021, 64, 470–480. [Google Scholar] [CrossRef]
- Rao, P.C.; Yoon, M. Potential liquid-organic hydrogen carrier (LOHC) systems: A review on recent progress. Energies 2020, 13, 6040. [Google Scholar] [CrossRef]
- Niermann, M.; Beckendorff, A.; Kaltschmitt, M.; Bonhoff, K. Liquid organic hydrogen carrier (LOHC)—Assessment based on chemical and economic properties. Int. J. Hydrogen Energy 2019, 44, 6631–6654. [Google Scholar] [CrossRef]
- Khamhaeng, P.; Laosiripojana, N.; Assabumrungrat, S.; Kim-Lohsoontorn, P. Techno-economic analysis of hydrogen production from dehydrogenation and steam reforming of ethanol for carbon dioxide conversion to methanol. Int. J. Hydrogen Energy 2021, 46, 30891–30902. [Google Scholar] [CrossRef]
- Bueno, J. Copper-Based Catalysts, Process for Preparing Same and Use Thereof. WO2004/080589 A2, 12 March 2004. [Google Scholar]
- Inui, K.K.; Takahashi, T.S.; Kurabayashi, T.I. Catalyst for Ester Production and Process for Producing Ester. U.S. Patent 2004/0242917 A1, 2 December 2004. [Google Scholar]
- Inui, K.K.; Takahashi, T.S.; Kurabayashi, T.I. Catalyst for Ester Production and Process for Producing Ester. U.S. Patent 7,091,155 B2, 15 August 2006. [Google Scholar]
- Colley, S.W.; Fawcett, C.R.; Sharif, M.; Tuck, M.W.M.; Watson, D.J.; Wood, M.A. Davy Process Tech., Republic of South Africa. Purification of Ethyl Acetate from Mixture Comprising Ethanol and Water by Pressure Swing Distillation. EP 1 117 629 B1, 15 December 2004. [Google Scholar]
- Colley, S.W.; Fawcett, C.R.; Rathmell, C.; Tuck, M.W.M. Process for the Preparation of Ethyl Acetate. EP 1 117 631 B1, 17 November 2004. [Google Scholar]
- Colley, S.W.; Fawcett, C.R.; Sharif, M.; Tuck, M.W.M.; Watson, D.J.; Wood, M.A. Process. U.S. Patent 7,553,397 B1, 30 June 2009. [Google Scholar]
- Santacesaria, E.; Di Serio, M.; Tesser, R.; Carotenuto, G. Process for the Production of Ethyl Acetate from Ethanol. WO2011/104738A2, 1 September 2011. [Google Scholar]
- Santacesaria, E.; Carotenuto, G.; Tesser, R.; Di Serio, M. Ethanol dehydrogenation to ethyl acetate by using copper and copper chromite catalysts. Chem. Eng. J. 2012, 179, 209–220. [Google Scholar] [CrossRef]
- Carotenuto, G.; Tesser, R.; Di Serio, M.; Santacesaria, E. Bioethanol as feedstock for chemicals such as acetaldehyde, ethyl acetate and pure hydrogen. Biomass Conv. Bioref. 2013, 3, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Santacesaria, E.; Carotenuto, G.; Tesser, R.; Di Serio, M. Production of pure hydrogen by ethanol dehydrogenation. Oil Gas Eur. Mag. 2011, 37, 99–102. [Google Scholar]
- Santacesaria, E.; Carotenuto, G.; Tesser, R.; Di Serio, M. Production of pure hydrogen by ethanol dehydrogenation. In Proceedings of the DGMK Conference, Berlin, Germany, 4–6 October 2010. [Google Scholar]
- Carotenuto, G.; Tesser, R.; Di Serio, M.; Santacesaria, E. Kinetic study of ethanol dehydrogenation to ethyl acetate promoted by a copper/copper chromite catalyst. Catal. Today 2013, 203, 202–210. [Google Scholar] [CrossRef]
- Luo, M.; Das, T.K.; Delibas, C.; Davis, B.H. Heterogeneous catalytic hydrogenation of ethyl acetate to produce ethanol. Top. Catal. 2014, 57, 757–761. [Google Scholar] [CrossRef]
- Di, W.; Cheng, J.; Tian, S.; Li, J.; Chen, J.; Sun, Q. Synthesis and characterization of supported copper phyllosilicate catalysts for acetic ester hydrogenation to ethanol. Appl. Catal. A Gen. 2016, 510, 244–259. [Google Scholar] [CrossRef]
- Zhu, Y.; Shi, L. Zn promoted Cu–Al catalyst for hydrogenation of ethyl acetate to alcohol. J. Ind. Eng. Chem. 2014, 20, 2341–2347. [Google Scholar] [CrossRef]
- Zhu, Y.; Shi, X.W.L. Hydrogenation of ethyl acetate to ethanol over bimetallic Cu-Zn/SiO2 catalysts prepared by means of coprecipitation. Bull. Korean Chem. Soc. 2014, 35, 141–146. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Yin, H.; Wang, A.; Hu, J.; Xue, W.; Yin, H.; Liu, S. Hydrogenation of ethyl acetate to ethanol over Cu/ZnO/MOx (MOx = SiO2, Al2O3, and ZrO2) catalysts. J. Ind. Eng. Chem. 2016, 37, 208–215. [Google Scholar] [CrossRef]
- Tran, B.L.; Johnson, S.I.; Brooks, K.P.; Autrey, S.T. Ethanol as a liquid organic hydrogen carrier for seasonal microgrid application: Catalysis, theory, and engineering feasibility. ACS Sustain. Chem. Eng. 2021, 9, 7130–7138. [Google Scholar] [CrossRef]
- Mevawala, C.; Brooks, K.; Bowden, M.E.; Breunig, H.M.; Tran, B.L.; Gutiérrez, O.Y.; Autrey, T.; Müller, K. The ethanol–ethyl acetate system as a biogenic hydrogen carrier. Energy Technol. 2022, 11, 2200892. [Google Scholar] [CrossRef]
- Stull, D.R.; Vestrum, E.F.; Sinke, G.C. The Chemical Thermodynamics of Organic Compounds, 1st ed.; John Wiley: New York, NY, USA, 1969. [Google Scholar]
- Finger, P.H.; Osmari, T.A.; Costa, M.S.; Bueno, J.M.C.; Gallo, J.M.R. The role of the interface between Cu and metal oxides in the ethanol dehydrogenation. Appl. Catal. A Gen. 2020, 589, 117236. [Google Scholar] [CrossRef]
- Pang, J.; Zheng, M.; Wang, C.; Liu, H.; Liu, X.; Sun, J.; Wang, Y.; Zhang, T. Hierarchical echinus-like Cu-MFI catalysts for ethanol dehydrogenation. ACS Catal. 2020, 10, 13624–13629. [Google Scholar] [CrossRef]
- Freitas, I.C.; Damyanova, S.; Oliveira, D.C.; Marquez, C.M.P.; Bueno, J.M.C. Effect of Cu content on the surface and catalytic properties of Cu/ZrO2 catalyst for ethanol dehydrogenation. J. Mol. Catal. A Chem. 2014, 381, 26–37. [Google Scholar] [CrossRef]
- Tu, Y.J.; Li, C.; Chen, Y.W. Effect of chromium promoter on copper catalysts in ethanol dehydrogenation. J. Chem. Technol. Biotechnol. 1994, 59, 141–147. [Google Scholar] [CrossRef]
- Men’shchikov, V.A.; Gol’dshtein, L.K.; Semenov, I.P. Kinetics of ethanol dehydrogenation into ethyl acetate. Kinet. Catal. 2014, 55, 12–17. [Google Scholar] [CrossRef]
- Carotenuto, G. Innovative Processes for the Production of Acetaldehyde, Ethyl Acetate and Pure Hydrogen by Ethanol. Ph.D. Thesis in Chemistry Macromolecular and Catalysis, University of Naples Federico II, Naples, Italy, November 2011. [Google Scholar]
- Private Communication: Davy Process Technology Report; Ethyl Acetate Process. 13 April 2004.
- Berg, L. Separation of Ethyl Acetate from Ethanol by Azeotropic Distillation. U.S. Patent 5,993,610 A, 30 November 1999. [Google Scholar]
- Zhu, Z.; Ri, Y.; Jia, H.; Wang, Y.; Wang, Y. Process evaluation on the separation of ethyl acetate ethanol using extractive distillation with ionic liquid. Sep. Purif. Technol. 2017, 181, 44–52. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, M.; Li, C.; Zeng, A. Design and control of extractive distillation process for separation of the minimum-boiling azeotrope ethyl-acetate and ethanol. Chem. Eng. Res. Des. 2018, 136, 57–70. [Google Scholar] [CrossRef]
- Gadewar, S.B. Ethyl Acetate Production. U.S. Patent 2012/0035390A1, 9 February 2012. [Google Scholar]
- Gadewar, S.B.; Vicente, B.C.; Norton, R.E.; Doherty, M.F. Ethyl Acetate Production. U.S. Patent 2013/0197266A1, 1 August 2013. [Google Scholar]
- Gadewar, S.B. Ethyl Acetate Production. U.S. Patent 8,562,921B2, 22 October 2013. [Google Scholar]
- Gadewar, S.B.; Stoimenov, P.K. Ethyl Acetate Production. U.S. Patent 2014/0012037A1, 9 January 2014. [Google Scholar]
- Gadewar, S.B.; Vicente, B.C.; Norton, R.E.; Doherty, M.F. Ethyl Acetate Production. U.S. Patent 9,079,851B2, 14 July 2015. [Google Scholar]
- Gadewar, S.B.; Stoimenov, P.K. Ethyl Acetate Production. U.S. Patent 9,447,018B2, 20 September 2016. [Google Scholar]
- Senior Design Reports of the University of Pennsylvania: Ethanol to Ethyl Acetate. Available online: https://core.ac.uk/download/pdf/219382692.pdf (accessed on 27 January 2023).
- Raich, B.A.; Foley, H.C. Ethanol dehydrogenation with a palladium membrane reactor: An alternative to Wacker chemistry. Ind. Eng. Chem. Res. 1998, 37, 3888–3895. [Google Scholar] [CrossRef]
- Keuler, J.N.; Lorenzen, L. Pd-Ag membranes and their application for dehydrogenation of ethanol in a membrane reactor. In Proceedings of the 221st National Meeting ACS, San Diego, CA, USA, 1–5 April 2001. [Google Scholar]
- Keuler, J.N.; Lorenzen, L. Comparing and modeling the dehydrogenation of ethanol in a plug-flow reactor and a Pd-Ag membrane reactor. Ind. Eng. Chem. Res. 2002, 41, 1960–1966. [Google Scholar] [CrossRef]
- Lin, W.H.; Chang, H.F. A study of ethanol dehydrogenation reaction in a palladium membrane reactor. Catal. Today 2004, 97, 181–188. [Google Scholar] [CrossRef]
- Sánchez, A.B.; Homs, N.; Miachon, S.; Dalmon, J.A.; Fierro, J.L.G.; de la Piscina, P.R. Direct transformation of ethanol into ethyl acetate through catalytic membranes containing Pd or Pd-Zn: Comparison with conventional supported catalysts. Green Chem. 2011, 13, 2569–2575. [Google Scholar] [CrossRef]
- Zeng, G.; Chen, T.; He, L.; Pinnau, I.; Lai, Z.; Huang, K.W. A green approach to ethyl acetate: Quantitative conversion of ethanol through direct dehydrogenation in a Pd-Ag membrane reactor. Chem. Eur. J. 2012, 18, 15940–15943. [Google Scholar] [CrossRef]
- Adkins, H.; Folkers, K. The catalytic hydrogenation of esters to alcohols. J. Am. Chem. Soc. 1931, 53, 1095–1097. [Google Scholar] [CrossRef]
- Thakur, D.S.; Carrick, W. Copper Chromite Hydrogenation Catalysts for Production of Fatty Alcohols. WO 2012/074841, 7 June 2012. [Google Scholar]
- Yuan, P.; Liu, Z.Y.; Zhang, W.; Sun, H.; Liu, S. Cu-Zn/Al2O3 catalyst for the hydrogenation of esters to alcohols. Chin. J. Catal. 2010, 31, 769–775. [Google Scholar] [CrossRef]
- He, L.; Cheng, H.; Liang, G.; Yu, Y.; Zhao, F. Effect of structure of CuO/ZnO/Al2O3 composites on catalytic performance for hydrogenation of fatty acid ester. Appl. Catal. A Gen. 2013, 452, 88–93. [Google Scholar] [CrossRef]
- Huang, H.; Cao, G.; Wang, S. An evaluation of alkylthiols and dialkyl disulfides on deactivation of Cu/Zn catalyst in hydrogenation of dodecyl methyl ester to dodecanol. J. Ind. Eng. Chem. 2014, 20, 988–993. [Google Scholar] [CrossRef]
- Brands, D.S.; Poels, E.K.; Bliek, A. Ester hydrogenolysis over promoted Cu/SiO2 catalysts. Appl. Catal. A Gen. 1999, 184, 279–289. [Google Scholar] [CrossRef]
- Kim, S.M.; Lee, M.E.; Choi, J.W.; Suh, D.J.; Suh, Y.W. Role of ZnO in Cu/ZnO/Al2O3 catalyst for hydrogenolysis of butyl butyrate. Catal. Commun. 2011, 12, 1328–1332. [Google Scholar] [CrossRef]
- Huang, X.; Ma, M.; Miao, S.; Zheng, Y.; Chen, M.; Shen, W. Hydrogenation of methyl acetate to ethanol over a highly stable Cu/SiO2 catalyst: Reaction mechanism and structural evolution. Appl. Catal. A Gen. 2017, 531, 79–88. [Google Scholar] [CrossRef]
- Zhao, Y.; Shan, B.; Wang, Y.; Zhou, J.; Wang, S.; Ma, X. An effective CuZn-SiO2 bimetallic catalyst prepared by hydrolysis precipitation method for the hydrogenation of methyl acetate to ethanol. Ind. Eng. Chem. Res. 2018, 57, 4526–4534. [Google Scholar] [CrossRef]
- Jiang, X.C.; Wang, Z.G.; Li, C.X. Intrinsic kinetics of ethyl acetate hydrogenolysis to ethanol over a Cu-Zn/Al2O3 catalyst. J. Beijing Univ. Chem. Technol. 2014, 41, 36–39. [Google Scholar]
- Schittkowski, J.; Tölle, K.; Anke, S.; Stürmer, S.; Muhler, M. On the bifunctional nature of Cu/ZrO2 catalysts applied in the hydrogenation of ethyl acetate. J. Catal. 2017, 352, 120–129. [Google Scholar] [CrossRef]
- Natal Santiago, M.A.; Sánchez-Castillo, M.A.; Cortight, R.D.; Dumesic, J.A. Catalytic reduction of acetic acid, methyl acetate, and ethyl acetate over silica-supported copper. J. Catal. 2000, 193, 16–28. [Google Scholar] [CrossRef]
- Niermann, M.; Drünert, S.; Kaltschmitt, M.; Bonhoff, K. Liquid organic hydrogen carriers (LOHCs)–techno-economic analysis of LOHCs in a defined process chain. Energy Environ. Sci. 2019, 12, 290–307. [Google Scholar] [CrossRef]
- Gallucci, F.; Basile, A. Pd-Ag membrane reactor for steam reforming reactions: A comparison between different fuels. Int. J. Hydrogen Energy 2008, 33, 1671–1687. [Google Scholar] [CrossRef]
- Rahimpour, M.R.; Samimi, F.; Babapoor, A.; Tohidian, T.; Mohebi, S. Palladium membranes applications in reaction systems for hydrogen separation and purification: A review. Chem. Eng. Process. 2017, 121, 24–49. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinetic Constants | Activation Energy (kJ/mol) | |
| k1-493K | 97.1 ± 6.8(mol/(gcat·h·atm)) | 151.67 ± 18.20 |
| k2-493K | 0.089 ± 9.8 × 10−3 (mol/(gcat·h·atm2)) | 54.18 ± 2.72 |
| k3-493K | 0.0011 ± 7.8 × 10−4 (mol/(gcat·h·atm2)) | 6.69 × 10−4 ± 7.53 × 10−5 |
| Adsorption parameters (atm−1) | Adsorption Enthalpy (kJ/mol) | |
| bEtOH-493K | 10.4 ± 0.83 | −106.82 ± 10.67 |
| bAcH-493K | 98.4 ± 12.79 | −29.37 ± 1.46 |
| bEA-493K | 41.2 ± 4.94 | −58.20 ± 0.59 |
| bH-493K | 2.5 × 10−4 ± 3.5 × 10−5 | −55.81 ± 6.15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santacesaria, E.; Tesser, R.; Fulignati, S.; Raspolli Galletti, A.M. The Perspective of Using the System Ethanol-Ethyl Acetate in a Liquid Organic Hydrogen Carrier (LOHC) Cycle. Processes 2023, 11, 785. https://doi.org/10.3390/pr11030785
Santacesaria E, Tesser R, Fulignati S, Raspolli Galletti AM. The Perspective of Using the System Ethanol-Ethyl Acetate in a Liquid Organic Hydrogen Carrier (LOHC) Cycle. Processes. 2023; 11(3):785. https://doi.org/10.3390/pr11030785
Chicago/Turabian StyleSantacesaria, Elio, Riccardo Tesser, Sara Fulignati, and Anna Maria Raspolli Galletti. 2023. "The Perspective of Using the System Ethanol-Ethyl Acetate in a Liquid Organic Hydrogen Carrier (LOHC) Cycle" Processes 11, no. 3: 785. https://doi.org/10.3390/pr11030785