
Immobilization of an Industrial β-Glucosidase from Aspergillus fumigatus and Its Use for Cellobiose Hydrolysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Enzyme Activity Assays
2.3. Protein Concentration Tests
2.4. Protein Sample Preparation and SDS-PAGE
2.5. Support Description and Activation for Protein Immobilization
2.6. Enzyme Immobilization Procedures
2.7. Preliminary Characterization of Immobilized Enzymes
2.8. Enzyme Activity on Cellobiose: Activity Tests
2.9. Enzyme Studies on Cellobiose: Kinetic Analysis
2.10. Enzyme Studies on Cellobiose: Operational Stability
3. Results and Discussion
3.1. Preliminary Characterisation of the Enzymatic Extracts
3.2. Enzyme Immobilization
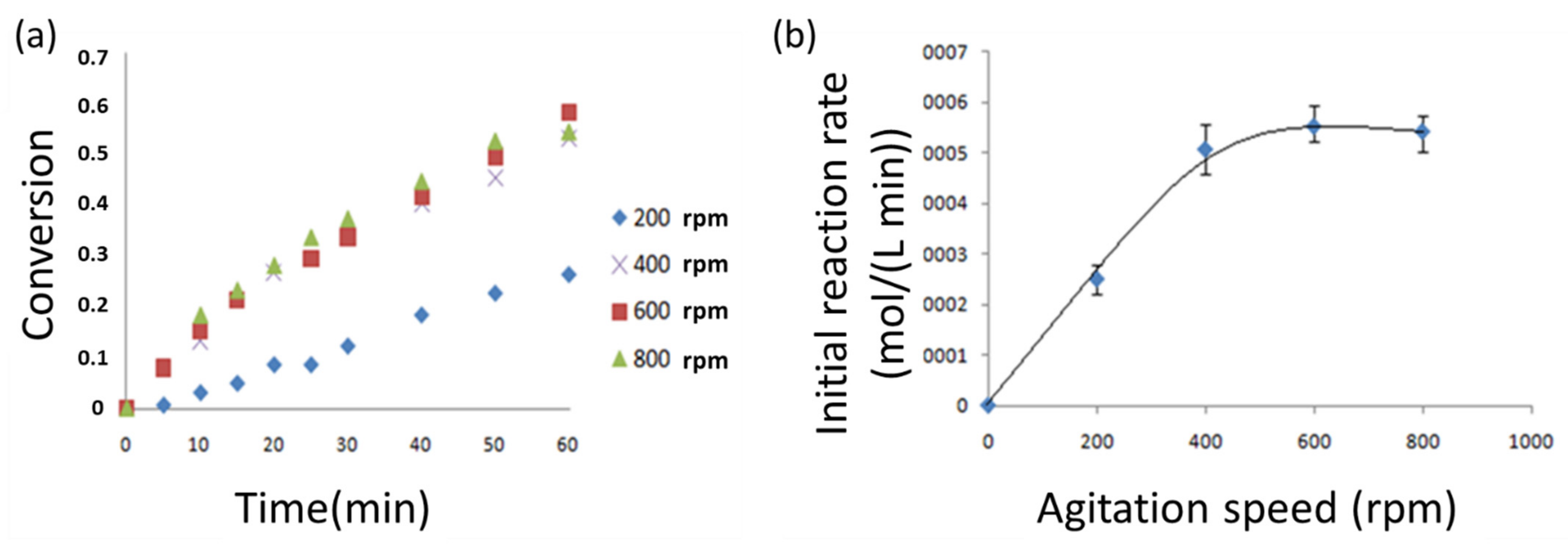
3.3. Cellobiose Hydrolysis: Preliminary Analysis
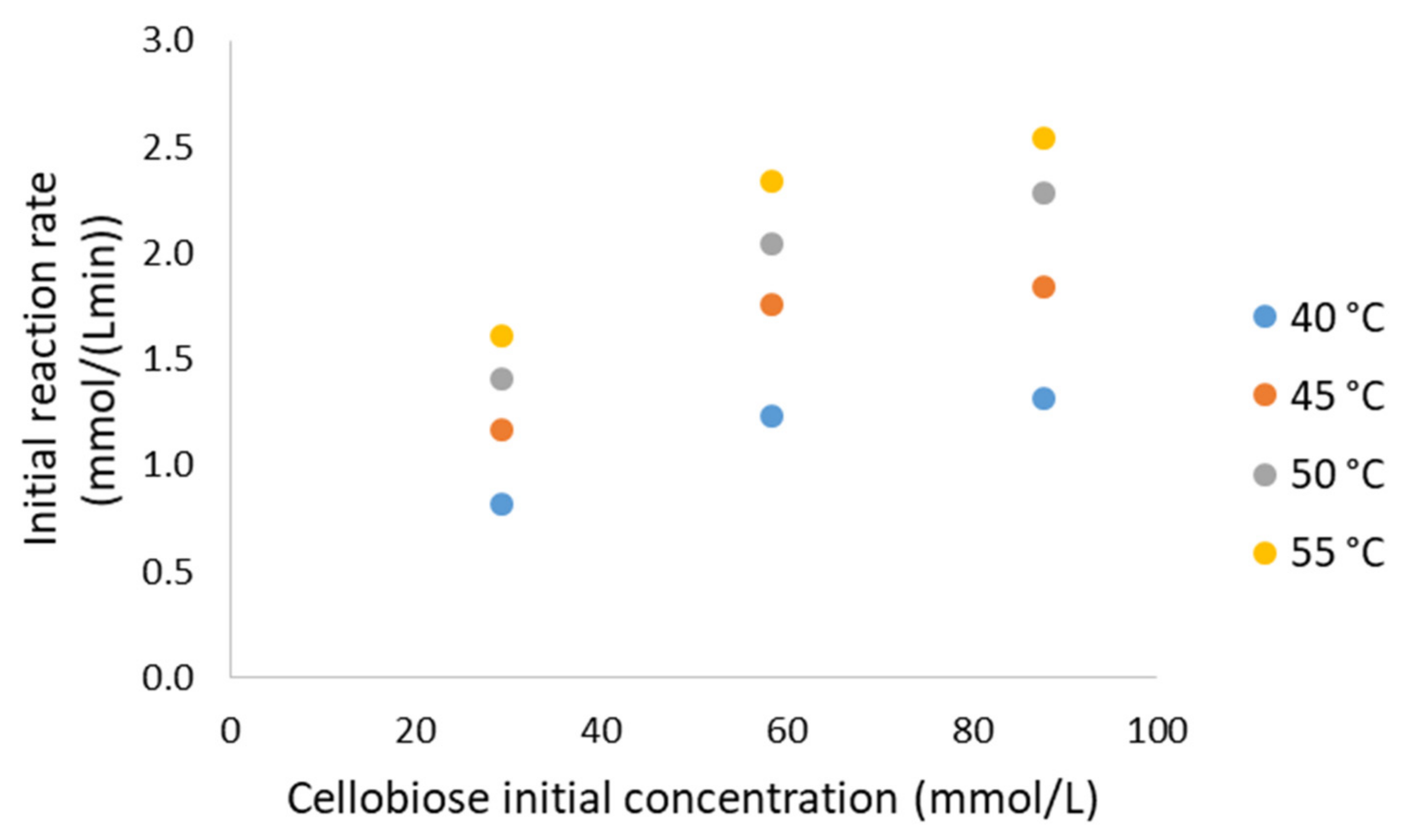
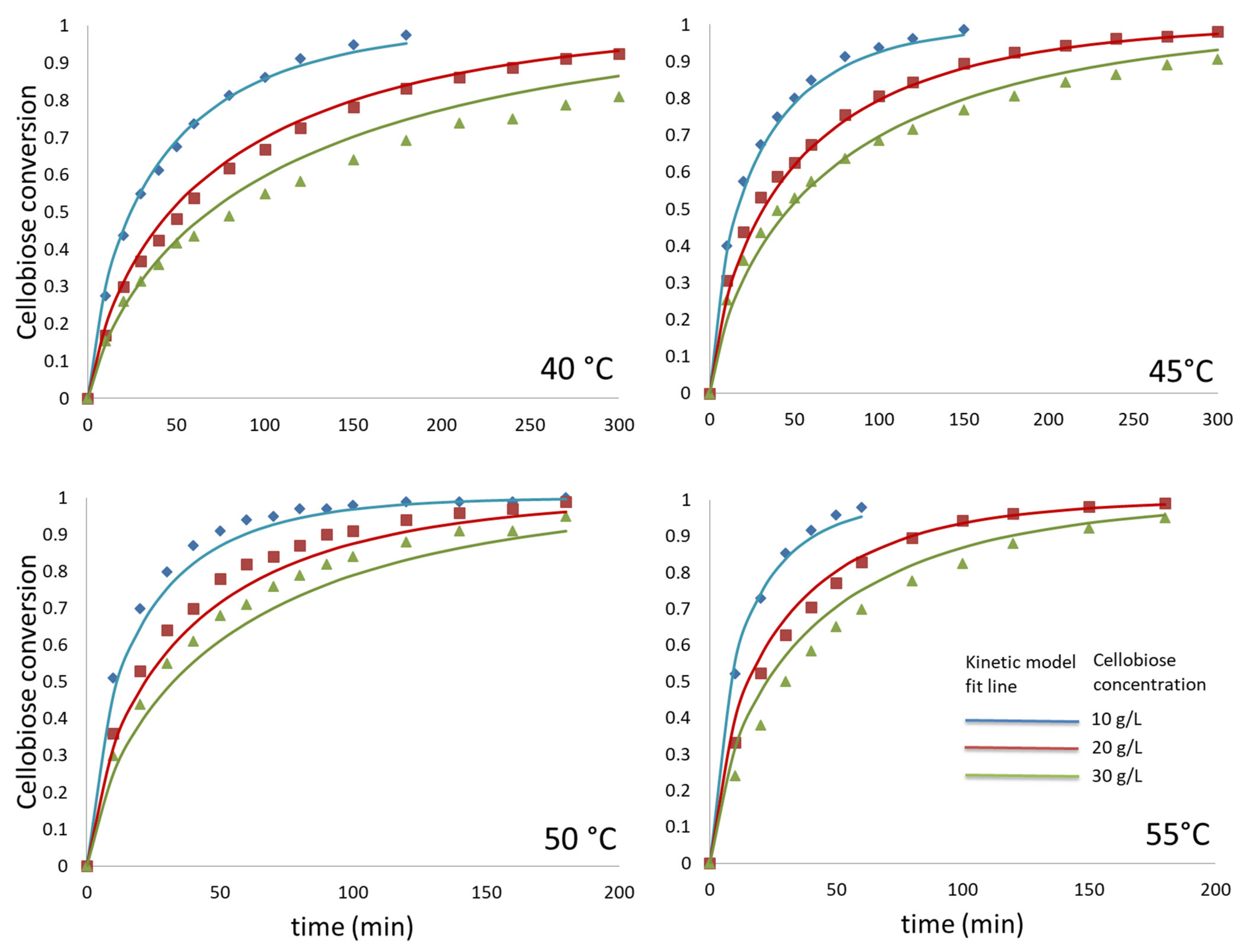
3.4. Cellobiose Hydrolysis: Kinetic Modelling
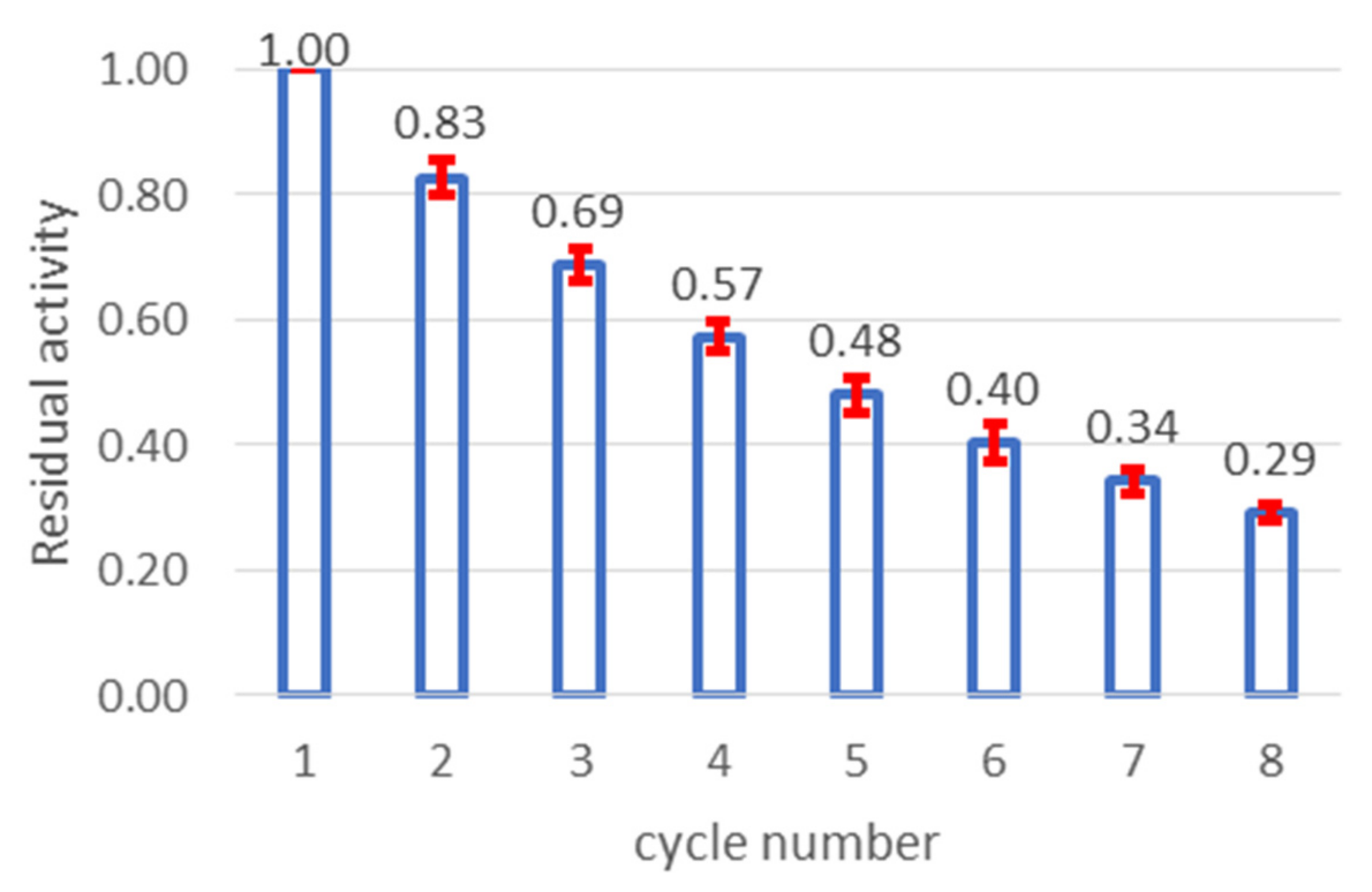
3.5. Immobilized Enzyme: Operational Stability
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Demirbaş, A. Biomass resource facilities and biomass conversion processing for fuels and chemicals. Energy Conv. Manag. 2001, 42, 1357–1378. [Google Scholar] [CrossRef]
- Bolton, J.R. Conversion and storage of solar energy. Ann. Rev. Energy 1979, 4, 353–401. [Google Scholar] [CrossRef]
- Calsavara, L.P.V.; Morales, F.F.; Zanin, G.M. Comparison of catalytic properties of free and immobilized cellobiase Novozym 188. Appl. Biochem. Biotechnol. 2001, 91–93, 615–626. [Google Scholar] [CrossRef]
- Agbor, V.B.; Cicek, N.; Sparling, R.; Berlin, A.; Levin, D.B. Biomass pretreatment: Fundamentals toward application. Biotechnol. Adv. 2011, 29, 675–685. [Google Scholar] [CrossRef]
- Chandel, A.K.; Chandrasekhar, G.; Silva, M.B.; da Silva, S.S. The realm of cellulases in biorefinery development. Crit. Rev. Biotechnol. 2012, 32, 187–202. [Google Scholar] [CrossRef]
- Srivastava, N.; Rathour, R.; Jha, S.; Pandey, K.; Srivastava, M.; Thakur, V.K.; Sengar, R.S.; Gupta, V.K.; Mazumder, P.B.; Khan, A.F.; et al. Microbial beta glucosidase enzymes: Recent advances in biomass conversation for biofuels application. Biomolecules 2019, 9, 220. [Google Scholar] [CrossRef] [Green Version]
- Singhania, R.R.; Patel, A.K.; Pandey, A.; Ganansounou, E. Genetic modification: A tool for enhancing beta-glucosidase production for biofuel application. Biores. Technol. 2017, 245, 1352–1361. [Google Scholar] [CrossRef]
- Huang, C.; Feng, Y.; Patel, G.; Xu, X.Q.; Qian, J.; Liu, Q.; Kai, G.Y. Production, immobilization and characterization of beta-glucosidase for application in cellulose degradation from a novel Aspergillus versicolor. Int. J. Biol. Macromol. 2021, 177, 437–446. [Google Scholar] [CrossRef]
- Vazquez-Ortega, P.G.; Alcaraz-Fructuoso, M.T.; Rojas-Contreras, J.A.; López-Miranda, J.; Fernandez-Lafuente, R. Stabilization of dimeric β-glucosidase from Aspergillus niger via glutaraldehyde immobilization under different conditions. Enz. Microb. Technol. 2018, 110, 38–45. [Google Scholar] [CrossRef]
- Nishida, V.S.; de Oliveira, R.F.; Brugnari, T.; Correa, R.C.G.; Peralta, R.A.; Castoldi, R.; de Souza, C.G.; Bracht, A.; Peralta, R.M. Immobilization of Aspergillus awamori β-glucosidase on commercial gelatin: An inexpensive and efficient process. Int. J. Biol. Macromol. 2018, 111, 1206–1213. [Google Scholar] [CrossRef]
- Wu, X.; Qu, B.; Liu, Y.; Ren, X.; Wang, S.; Quan, Y. Highly enhanced activity and stability via affinity induced immobilization β-glucosidase from Aspergillus niger onto amino-based silica for the biotransformation of ginsenoside Rb1. J. Chromatog. A 2021, 1653, 462388. [Google Scholar] [CrossRef]
- Tu, M.; Zhang, X.; Kurabi, A.; Gilkes, N.; Mabee, W.; Saddler, J. Immobilization of β-glucosidase on Eupergit C for lignocellulose hydrolysis. Biotechnol. Lett. 2006, 28, 151–156. [Google Scholar] [CrossRef]
- Silva, T.M.; Pessela, B.C.; Silva, J.C.R.; Lima, M.S.; Jorge, J.A.; Guisan, J.M.; Polizeli, M.L.T.M. Immobilization and high stability of an extracellular β-glucosidase from Aspergillus japonicus by ionic interactions. J. Mol. Catal. B Enzym. 2014, 104, 95–100. [Google Scholar] [CrossRef]
- Alvarez-Gonzalez, C.; Santos, V.E.; Ladero, M.; Bolivar, J.M. Immobilization-Stabilization of β-Glucosidase for Implementation of Intensified Hydrolysis of Cellobiose in Continuous Flow Reactors. Catalysts 2022, 12, 80. [Google Scholar] [CrossRef]
- Katchalski-Katzir, E.; Kraemer, D.M. Eupergit® C, a carrier for immobilization of enzymes of industrial potential. J. Mol. Catal. B Enzym. 2000, 10, 157–176. [Google Scholar] [CrossRef]
- Hilterhaus, L.; Minow, B.; Müller, J.; Berheide, M.; Quitmann, H.; Katzer, M.; Thum, O.; Antranikian, G.; Zeng, A.P.; Liese, A. Practical application of different enzymes immobilized on Sepabeads. Bioproc. Biosys. Eng. 2008, 31, 163–171. [Google Scholar] [CrossRef]
- Mateo, C.; Abian, O.; Fernández-Lorente, G.; Pedroche, J.; Fernández-Lafuente, R.; Guisan, J.M.; Tam, A.; Daminiti, M. Epoxy sepabeads: A novel epoxy support for stabilization of industrial enzymes via very intense multipoint covalent attachment. Biotechnol. Progr. 2002, 18, 629–634. [Google Scholar] [CrossRef]
- Kahar, U.M.; Sani, M.H.; Chan, K.-G.; Goh, K.M. Immobilization of α-amylase from Anoxybacillus sp. SK3-4 on ReliZyme and immobead supports. Molecules 2016, 21, 1196. [Google Scholar] [CrossRef] [Green Version]
- Bu, Y.; Zhang, T.; Jiang, B.; Chen, J. Improved performance of d-psicose 3-epimerase by immobilisation on amino-epoxide support with intense multipoint attachment. Foods 2021, 10, 831. [Google Scholar] [CrossRef]
- Rashid, M.H.; Siddiqui, K.S. Carboxy-group modification: High-temperature activation of charge-neutralized and charge-reversed beta-glucosidases from Aspergillus niger. Biotechnol. Appl. Biochem. 1998, 27, 231–237. [Google Scholar]
- Bezbradica, D.I.; Mateo, C.; Guisan, J.M. Novel support for enzyme immobilization prepared by chemical activation with cysteine and glutaraldehyde. J. Mol. Catal. B Enzym. 2014, 102, 218–224. [Google Scholar] [CrossRef]
- Bradford, M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 277, 680–685. [Google Scholar] [CrossRef]
- Lima, M.A.; Oliveira-Neto, M.; Kadowaki, M.A.S.; Rosseto, F.R.; Prates, E.T.; Squina, F.M.; Leme, A.F.P.; Skaf, M.S.; Polikarpov, I. Aspergillus niger β-glucosidase has a cellulase-like tadpole molecular shape: Insights into glycoside hydrolase family 3 (GH3) β-glucosidase structure and function. J. Biol. Chem. 2013, 288, 32991–33005. [Google Scholar] [CrossRef] [Green Version]
- Wojtusik, M.; Yepes, C.M.; Villar, J.C.; Cordes, A.; Arroyo, M.; Garcia-Ochoa, F.; Ladero, M. Kinetic modeling of cellobiose by a β-glucosidase from Aspergillus fumigatus. Chem. Eng. Res. Design 2018, 136, 502–512. [Google Scholar] [CrossRef]
- Wojtusik, M.; Villar, J.C.; Ladero, M.; Garcia-Ochoa, F. Physico-chemical kinetic modelling of hydrolysis of a steam-explosion pre-treated corn stover: A two-step approach. Bioresour. Technol. 2018, 268, 592–598. [Google Scholar] [CrossRef]
- Sheldon, R.A. Enzyme immobilization: The quest for optimum performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- López-Gallego, F.; Montes, T.; Fuentes, M.; Alonso, N.; Grazu, V.; Betancor, L.; Guisán, J.M.; Fernández-Lafuente, R. Improved stabilization of chemically aminated enzymes via multipoint covalent attachment on glyoxyl supports. J. Biotechnol. 2005, 116, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Betancor, L.; López-Gallego, F.; Hidalgo, A.; Alonso-Morales, N.; Mateo, G.D.O.C.; Fernández-Lafuente, R.; Guisán, J.M. Different mechanisms of protein immobilization on glutaraldehyde activated supports: Effect of support activation and immobilization conditions. Enz. Microbiol. Technol. 2006, 39, 877–882. [Google Scholar] [CrossRef]
- Mallin, H.; Wulf, H.; Bornscheuer, U.T. A self-sufficient Baeyer–Villiger biocatalysis system for the synthesis of ɛ-caprolactone from cyclohexanol. Enzyme Microb. Technol. 2013, 53, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Le Traon-Masson, M.P.; Pellerin, P. Purification and characterization or two β-D-glucosidases from an Aspergillus niger enzyme preparation: Affinity and specificity toward glucosylated compounds of the processing of fruits. Enzyme Microb. Technol. 1998, 22, 374–382. [Google Scholar] [CrossRef]
- Nagy, F.; Gyujto, I.; Tasnádi, G.; Barna, B.; Balogh-Weiser, D.; Faber, K.; Poppe, L.; Hall, M. Design and application of a bi-functional redox biocatalyst through covalent co-immobilization of ene-reductase and glucose dehydrogenase. J. Biotechnol. 2020, 323, 246–253. [Google Scholar] [CrossRef]
- Bilgin, R.; Yalcin, M.S.; Yildirim, D. Optimization of covalent immobilization of Trichoderma reesei cellulase onto modified ReliZyme HA403 and Sepabeads EC-EP supports for cellulose hydrolysis, in buffer and ionic liquids/buffer media. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1276–1284. [Google Scholar] [CrossRef]
- D’Arrigo, P.; Allegretti, C.; Fiorati, A.; Piubelli, L.; Rosini, E.; Tessaro, D.; Valentino, M.; Pollegioni, L. Immobilization of L-aspartate oxidase from Sulfolobus tokodaii as a biocatalyst for resolution of aspartate solutions. Catal. Sci. Technol. 2015, 5, 1106–1114. [Google Scholar] [CrossRef]
- Das, A.; Paul, T.; Ghosh, P.; Halder, S.K.; Das Mohapatra, P.K.; Pati, B.R.; Mondal, K.C. Kinetic study of a glucose tolerant β-glucosidase from Aspergillus fumigatus ABK9 entrapped into alginate beads. Waste Biomass Valor. 2015, 6, 53–61. [Google Scholar] [CrossRef]
- Wojtusik, M.; Vergara, P.; Villar, J.C.; Garcia-Ochoa, F.; Ladero, M. Thermal and operational deactivation of Aspergillus fumigatus β-glucosidase in ethanol/water pretreated wheat straw enzymatic hydrolysis. J. Biotechnol. 2019, 292, 32–38. [Google Scholar] [CrossRef]
- Fathi, Z.; Doustkhah, E.; Ebrahimipour, G.; Darvishi, F. Noncovalent immobilization of Yarrowia lipolytica lipase on dendritic-like amino acid-functionalized silica nanoparticles. Biomolecules 2019, 9, 502. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immobilized Preparation | Selected Support | Enzyme Cocktail | Immobilization Process Time | aL | YL | YS | aS |
|---|---|---|---|---|---|---|---|
| RN1 | ReliZyme™ EP403 | Novozym 188 | 24 h | 8.6 | 24.7% | 0.18% | 0.04 |
| RN2 | ReliZyme™ EP403 | Novozym 188 | 72 h | 2.7 | 28.9% | 11.9% | 0.9 |
| RN3 | ReliZyme™ HA403-GA | Novozym 188 | 24 h | 2.7 | 48.5% | 5% | 0.7 |
| RN4 | ReliZyme™ HA403-GA | Novozym 188 | 24 h | 1.4 | 35.7% | 54.8% | 3.3 |
| RN5 | Eupergit® C | Novozym 188 | 72 h | 2.4 | 14.3% | 2.8% | 0.09 |
| RN6 | Sepabeads™ EC-EP303 | Novozym 188 | 72 h | 2.4 | 5.7% | 17.9% | 0.2 |
| RN7 | ReliZyme™ HA403-GA | Novozym 188 | 24 h | 0.45 | 71% | 46% | 1.5 |
| RN8 | ReliZyme™ HA403-GA | ASA-1000 | 24 h | 0.8 | 100% | 73.8% | 6.2 |
| Immobilized Preparation | aL | YL | YS | aS | YScel | aScel |
|---|---|---|---|---|---|---|
| RN8 | 0.8 | 100% | 73.8% | 6.2 | 94.2% | 3.5 |
| RN9 | 1.6 | 100% | 71.8% | 11.5 | 91.4% | 6.8 |
| RN10 | 3.2 | 100% | 67.4% | 21.6 | 92.1% | 13.7 |
| RN11 | 6.4 | 100% | 63.2% | 40.4 | 89.5% | 26.6 |
| RN12 | 12.8 | 100% | 58.4% | 74.8 | 90.2% | 53.6 |
| RN13 | 25.6 | 100% | 45.1% | 116 | 87.3% | 103.8 |
| RN14 | 51.2 | 80% | 37.3% | 191 | 79.5% | 189.0 |
| RN15 | 102.4 | 45% | 33.4% | 342 | 68.3% | 324.8 |
| Kinetic Model | Ln k10 | Ea/R (k1) | KM | KI | SQR | RMSE | F-Value |
|---|---|---|---|---|---|---|---|
| M1 n = 1 | 16.26 ± 2.45 | 7632 ± 536 | 0.0025 ± 0.008 | 0.006 ± 0.002 | 0.198 | 0.0392 | 13,879 |
| M2 n = 2 | 15.78 ± 0.71 | 7516 ± 221 | 0.020 ± 0.003 | 0.042 ± 0.003 | 0.193 | 0.0378 | 14,389 |
| M3 n = 3 | 15.72 ± 0.72 | 7493 ± 225 | 0.025 ± 0.004 | 0.091 ± 0.005 | 0.202 | 0.0384 | 13,742 |
| M4 n = 4 | 15.69 ± 0.73 | 7478 ± 227 | 0.028 ± 0.004 | 0.142 ± 0.006 | 0.207 | 0.0393 | 13,320 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yepes, C.; Estévez, J.; Arroyo, M.; Ladero, M. Immobilization of an Industrial β-Glucosidase from Aspergillus fumigatus and Its Use for Cellobiose Hydrolysis. Processes 2022, 10, 1225. https://doi.org/10.3390/pr10061225
Yepes C, Estévez J, Arroyo M, Ladero M. Immobilization of an Industrial β-Glucosidase from Aspergillus fumigatus and Its Use for Cellobiose Hydrolysis. Processes. 2022; 10(6):1225. https://doi.org/10.3390/pr10061225
Chicago/Turabian StyleYepes, Clara, Juliana Estévez, Miguel Arroyo, and Miguel Ladero. 2022. "Immobilization of an Industrial β-Glucosidase from Aspergillus fumigatus and Its Use for Cellobiose Hydrolysis" Processes 10, no. 6: 1225. https://doi.org/10.3390/pr10061225