
Automation of Modeling and Calibration of Integrated Preparative Protein Chromatography Systems


Abstract
:1. Introduction
2. Theory
2.1. Mathematical Modeling
2.2. Yamamoto Method
3. Materials and Methods
3.1. Materials
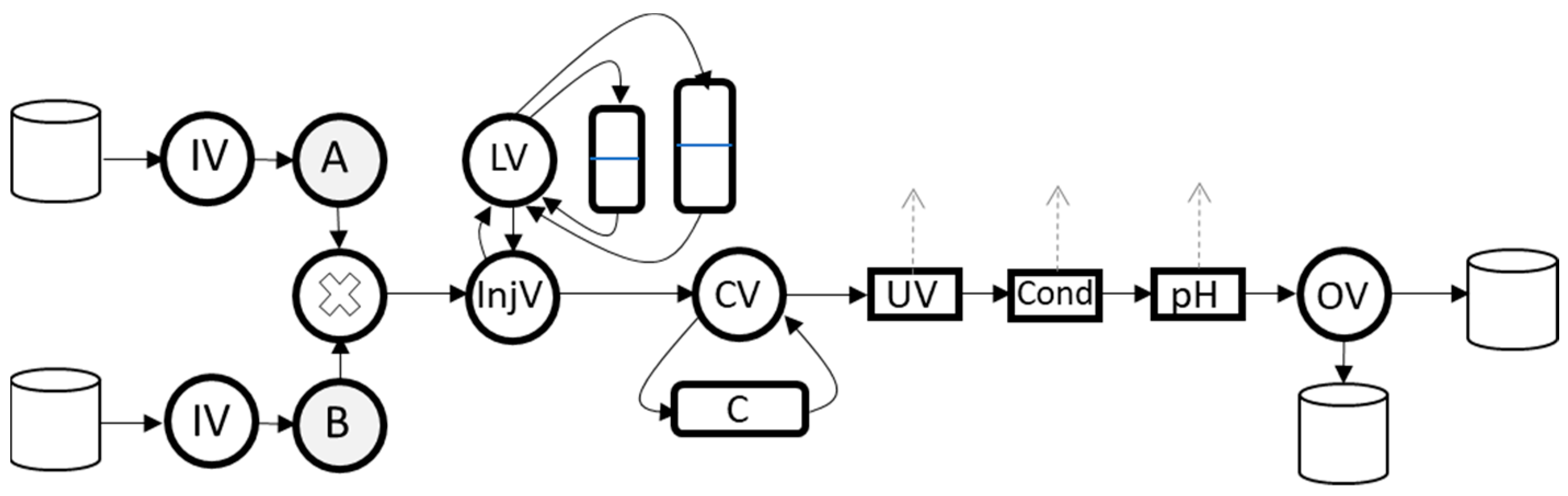
3.2. Experimental Setup
3.3. Experiments
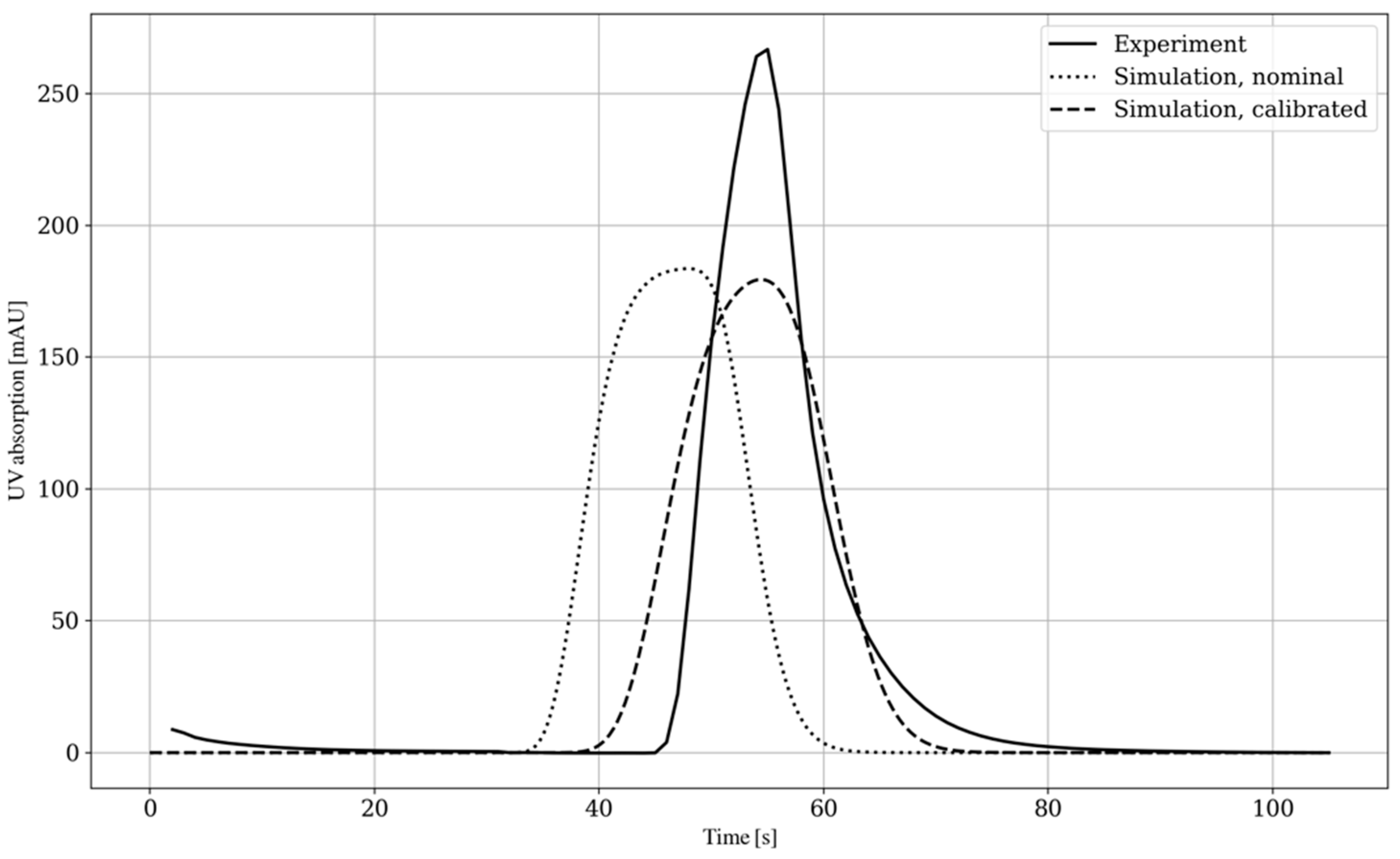
3.3.1. By-Pass Experiment (I)
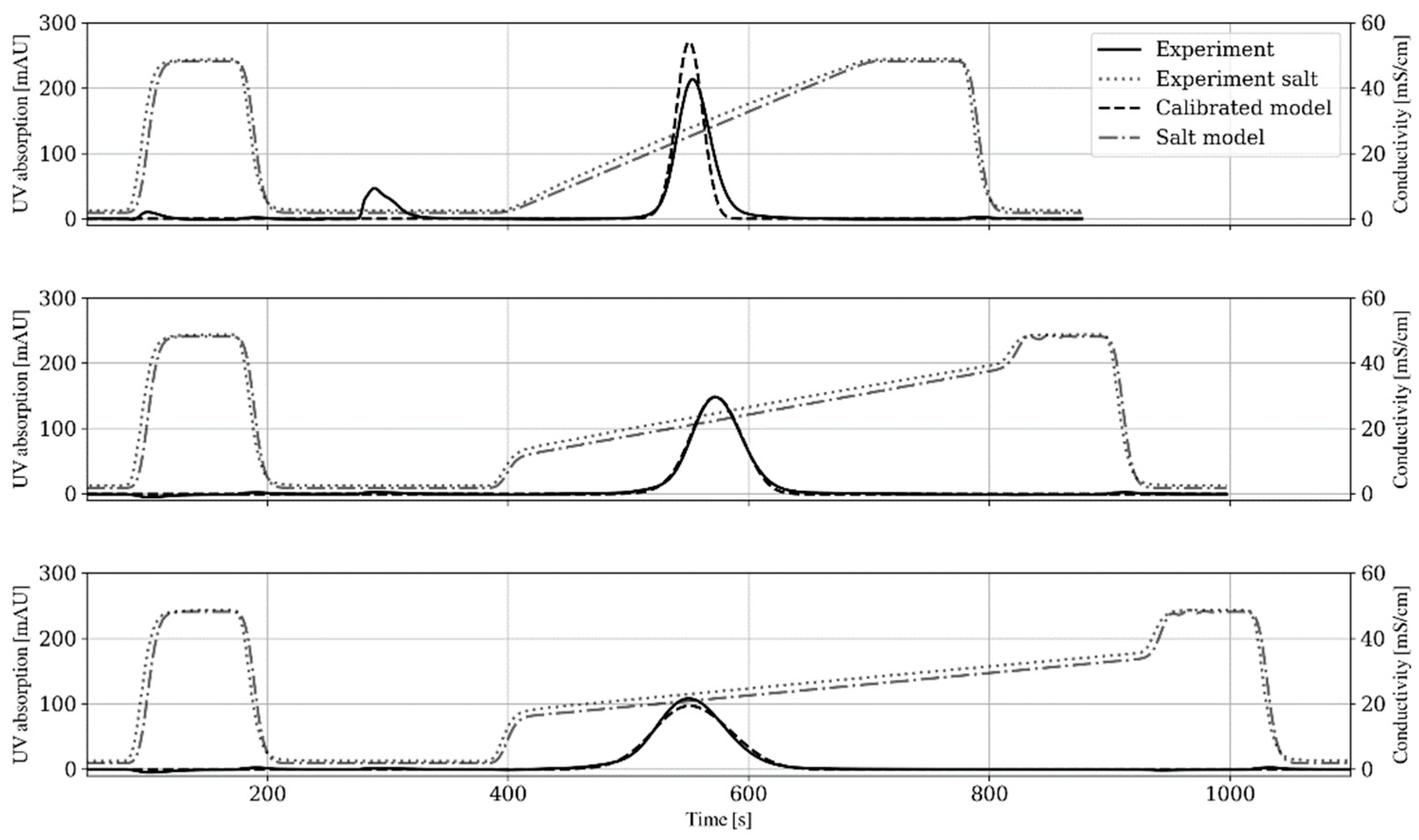
3.3.2. Packed Bed Experiment (II)
3.3.3. Linear Gradient Experiment (III)
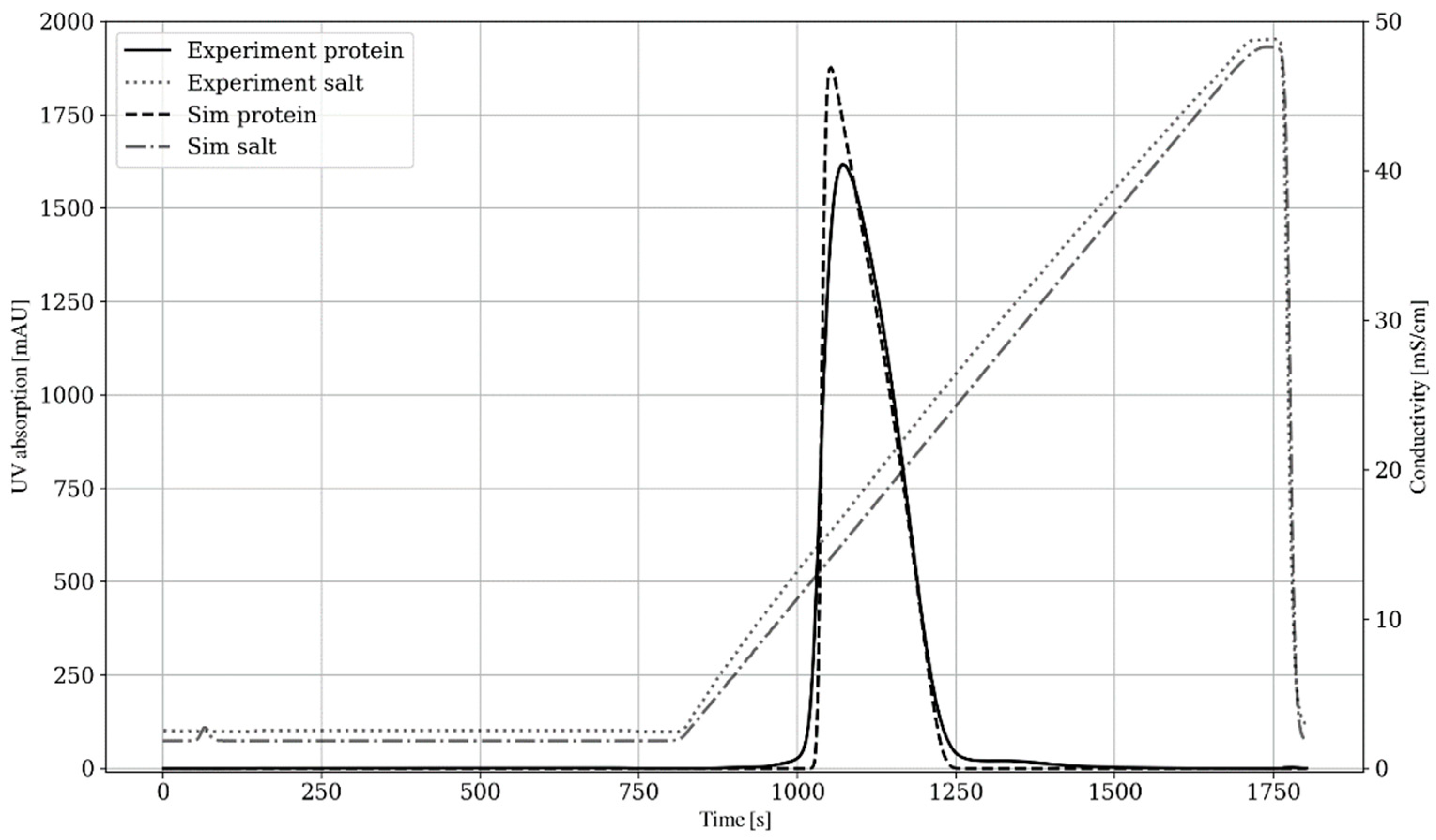
3.3.4. Overloading Experiment (IV)
3.4. Orbit
3.4.1. External Controller for Physical Experiments
3.4.2. Automatically Generated Simulator
3.4.3. Model Calibration
4. Results and Discussion
4.1. By-Pass Modeling and Calibration
4.2. Packed Bed Modeling and Calibration
4.3. Linear Gradient Adsorption Modeling and Calibration
4.4. Overloaded Adsorption Modeling and Calibration
4.5. General Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farid, S.S. Process Economics of Industrial Monoclonal Antibody Manufacture. J. Chromatogr. B 2007, 848, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Konstantinov, K.B.; Cooney, C.L. White Paper on Continuous Bioprocessing May 20–21 2014 Continuous Manufacturing Symposium. J. Pharm. Sci. 2015, 104, 813–820. [Google Scholar] [CrossRef]
- Jungbauer, A. Continuous Downstream Processing of Biopharmaceuticals. Trends Biotechnol. 2013, 31, 479–492. [Google Scholar] [CrossRef]
- Narayanan, H.; Sponchioni, M.; Morbidelli, M. Integration and Digitalization in the Manufacturing of Therapeutic Proteins. Chem. Eng. Sci. 2022, 248, 117159. [Google Scholar] [CrossRef]
- Degerman, M.; Westerberg, K.; Nilsson, B. A Model-Based Approach to Determine the Design Space of Preparative Chromatography. Chem. Eng. Technol. 2009, 32, 1195–1202. [Google Scholar] [CrossRef]
- Sellberg, A.; Holmqvist, A.; Magnusson, F.; Andersson, C.; Nilsson, B. Discretized Multi-Level Elution Trajectory: A Proof-of-Concept Demonstration. J. Chromatogr. A 2017, 1481, 73–81. [Google Scholar] [CrossRef]
- Guélat, B.; Ströhlein, G.; Lattuada, M.; Delegrange, L.; Valax, P.; Morbidelli, M. Simulation Model for Overloaded Monoclonal Antibody Variants Separations in Ion-Exchange Chromatography. J. Chromatogr. A 2012, 1253, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Close, E.J.; Salm, J.R.; Bracewell, D.G.; Sorensen, E. Modelling of Industrial Biopharmaceutical Multicomponent Chromatography. Chem. Eng. Res. Des. 2014, 92, 1304–1314. [Google Scholar] [CrossRef] [Green Version]
- Gomis-Fons, J.; Schwarz, H.; Zhang, L.; Andersson, N.; Nilsson, B.; Castan, A.; Solbrand, A.; Stevenson, J.; Chotteau, V. Model-Based Design and Control of a Small-Scale Integrated Continuous End-to-End MAb Platform. Biotechnol. Prog. 2020, 36, e2995. [Google Scholar] [CrossRef]
- Silva, R.J.S.; Rodrigues, R.C.R.; Osuna-Sanchez, H.; Bailly, M.; Valéry, E.; Mota, J.P.B. A New Multicolumn, Open-Loop Process for Center-Cut Separation by Solvent-Gradient Chromatography. J. Chromatogr. A 2010, 1217, 8257–8269. [Google Scholar] [CrossRef]
- Nilsson, B.; Andersson, N. Simulation of Process Chromatography. In Preparative Chromatography for Separation of Proteins; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 81–110. ISBN 978-1-119-03111-6. [Google Scholar]
- Rosen, R.; von Wichert, G.; Lo, G.; Bettenhausen, K.D. About The Importance of Autonomy and Digital Twins for the Future of Manufacturing. IFAC-Papersonline 2015, 48, 567–572. [Google Scholar] [CrossRef]
- Kritzinger, W.; Karner, M.; Traar, G.; Henjes, J.; Sihn, W. Digital Twin in Manufacturing: A Categorical Literature Review and Classification. IFAC-Papersonline 2018, 51, 1016–1022. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, O.; Sampat, C.; Bhalode, P.; Ramachandran, R.; Ierapetritou, M. Digital Twins in Pharmaceutical and Biopharmaceutical Manufacturing: A Literature Review. Processes 2020, 8, 1088. [Google Scholar] [CrossRef]
- Grossmann, C.; Ströhlein, G.; Morari, M.; Morbidelli, M. Optimizing Model Predictive Control of the Chromatographic Multi-Column Solvent Gradient Purification (MCSGP) Process. J. Process Control 2010, 20, 618–629. [Google Scholar] [CrossRef]
- Mouellef, M.; Vetter, F.L.; Zobel-Roos, S.; Strube, J. Fast and Versatile Chromatography Process Design and Operation Optimization with the Aid of Artificial Intelligence. Processes 2021, 9, 2121. [Google Scholar] [CrossRef]
- Persson, P.; Kempe, H.; Zacchi, G.; Nilsson, B. A Methodology for Estimation of Mass Transfer Parameters in a Detailed Chromatography Model Based on Frontal Experiments. Chem. Eng. Res. Des. 2004, 82, 517–526. [Google Scholar] [CrossRef]
- Kumar, V.; Leweke, S.; von Lieres, E.; Rathore, A.S. Mechanistic Modeling of Ion-Exchange Process Chromatography of Charge Variants of Monoclonal Antibody Products. J. Chromatogr. A 2015, 1426, 140–153. [Google Scholar] [CrossRef]
- Brooks, C.A.; Cramer, S.M. Steric Mass-Action Ion Exchange: Displacement Profiles and Induced Salt Gradients. AIChE J. 1992, 38, 1969–1978. [Google Scholar] [CrossRef]
- Karlsson, D.; Jakobsson, N.; Brink, K.-J.; Axelsson, A.; Nilsson, B. Methodologies for Model Calibration to Assist the Design of a Preparative Ion-Exchange Step for Antibody Purification. J. Chromatogr. A 2004, 1033, 71–82. [Google Scholar] [CrossRef]
- Jakobsson, N.; Degerman, M.; Stenborg, E.; Nilsson, B. Model Based Robustness Analysis of an Ion-Exchange Chromatography Step. J. Chromatogr. A 2007, 1138, 109–119. [Google Scholar] [CrossRef]
- von Lieres, E.; Andersson, J. A Fast and Accurate Solver for the General Rate Model of Column Liquid Chromatography. Comput. Chem. Eng. 2010, 34, 1180–1191. [Google Scholar] [CrossRef]
- Borg, N.; Brodsky, Y.; Moscariello, J.; Vunnum, S.; Vedantham, G.; Westerberg, K.; Nilsson, B. Modeling and Robust Pooling Design of a Preparative Cation-Exchange Chromatography Step for Purification of Monoclonal Antibody Monomer from Aggregates. J. Chromatogr. A 2014, 1359, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.; Larsen, T.; Todd, R.J. Modeling Preparative Cation Exchange Chromatography of Monoclonal Antibodies. In Preparative Chromatography for Separation of Proteins; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 399–427. ISBN 978-1-119-03111-6. [Google Scholar]
- Saleh, D.; Wang, G.; Müller, B.; Rischawy, F.; Kluters, S.; Studts, J.; Hubbuch, J. Straightforward Method for Calibration of Mechanistic Cation Exchange Chromatography Models for Industrial Applications. Biotechnol. Prog. 2020, 36, e2984. [Google Scholar] [CrossRef] [PubMed]
- Osberghaus, A.; Hepbildikler, S.; Nath, S.; Haindl, M.; von Lieres, E.; Hubbuch, J. Determination of Parameters for the Steric Mass Action Model—A Comparison between Two Approaches. J. Chromatogr. A 2012, 1233, 54–65. [Google Scholar] [CrossRef]
- Hahn, T.; Baumann, P.; Huuk, T.; Heuveline, V.; Hubbuch, J. UV Absorption-Based Inverse Modeling of Protein Chromatography. Eng. Life Sci. 2016, 16, 99–106. [Google Scholar] [CrossRef]
- Huuk, T.C.; Hahn, T.; Osberghaus, A.; Hubbuch, J. Model-Based Integrated Optimization and Evaluation of a Multi-Step Ion Exchange Chromatography. Sep. Purif. Technol. 2014, 136, 207–222. [Google Scholar] [CrossRef]
- Raje, P.; Pinto, N.G. Combination of the Steric Mass Action and Non-Ideal Surface Solution Models for Overload Protein Ion-Exchange Chromatography. J. Chromatogr. A 1997, 760, 89–103. [Google Scholar] [CrossRef]
- Müller-Späth, T.; Ströhlein, G.; Aumann, L.; Kornmann, H.; Valax, P.; Delegrange, L.; Charbaut, E.; Baer, G.; Lamproye, A.; Jöhnck, M.; et al. Model Simulation and Experimental Verification of a Cation-Exchange IgG Capture Step in Batch and Continuous Chromatography. J. Chromatogr. A 2011, 1218, 5195–5204. [Google Scholar] [CrossRef]
- Ojala, F.; Max-Hansen, M.; Kifle, D.; Borg, N.; Nilsson, B. Modelling and Optimisation of Preparative Chromatographic Purification of Europium. J. Chromatogr. A 2012, 1220, 21–25. [Google Scholar] [CrossRef]
- Max-Hansen, M.; Knutson, H.-K.; Jönsson, C.; Degerman, M.; Nilsson, B. Modeling Preparative Chromatographic Separation of Heavy Rare Earth Elements and Optimization of Thulium Purification. Adv. Mater. Phys. Chem. 2015, 5, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Rüdt, M.; Gillet, F.; Heege, S.; Hitzler, J.; Kalbfuss, B.; Guélat, B. Combined Yamamoto Approach for Simultaneous Estimation of Adsorption Isotherm and Kinetic Parameters in Ion-Exchange Chromatography. J. Chromatogr. A 2015, 1413, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Kadoya, T.; Yoshida, H.; Tamada, T.; Yamamoto, S. Rational Methods for Predicting Human Monoclonal Antibodies Retention in Protein A Affinity Chromatography and Cation Exchange Chromatography: Structure-Based Chromatography Design for Monoclonal Antibodies. J. Chromatogr. A 2005, 1093, 126–138. [Google Scholar] [CrossRef] [PubMed]
- CaptoTM. SP ImpRes Capto Q ImpRes. 2014. Available online: https://cdn.cytivalifesciences.com/api/public/content/digi-37317-pdf (accessed on 29 March 2022).
- Andersson, N. The Orbit Controller; Department of Chemical Engineering, Lund University: Lund, Sweden, 2018. [Google Scholar]
- Leweke, S.; von Lieres, E. Chromatography Analysis and Design Toolkit (CADET). Comput. Chem. Eng. 2018, 113, 274–294. [Google Scholar] [CrossRef]
- Scipy.Integrate.Solve_ivp—SciPy v1.8.0 Manual. Available online: https://docs.scipy.org/doc/scipy/reference/generated/scipy.integrate.solve_ivp.html (accessed on 10 March 2022).
- Felinger, A. Peak Detection. Data Anal. Signal Process. Chromatogr. 1998, 21, 184–190. [Google Scholar]
- Scipy.Optimize.Minimize—SciPy v1.6.0 Reference Guide. Available online: https://docs.scipy.org/doc/scipy-1.6.0/reference/generated/scipy.optimize.minimize.html#scipy.optimize.minimize (accessed on 2 May 2022).
- Heymann, W.; Glaser, J.; Schlegel, F.; Johnson, W.; Rolandi, P.; von Lieres, E. Advanced Score System and Automated Search Strategies for Parameter Estimation in Mechanistic Chromatography Modeling. J. Chromatogr. A 2022, 1661, 462693. [Google Scholar] [CrossRef]
- Samuelsson, J.; Edström, L.; Forssén, P.; Fornstedt, T. Injection Profiles in Liquid Chromatography. I. A Fundamental Investigation. J. Chromatogr. A 2010, 1217, 4306–4312. [Google Scholar] [CrossRef]
- Karlsson, D.; Jakobsson, N.; Axelsson, A.; Nilsson, B. Model-Based Optimization of a Preparative Ion-Exchange Step for Antibody Purification. J. Chromatogr. A 2004, 1055, 29–39. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calibration Step | Experiment | Parameters | Objective Function (SSE) |
|---|---|---|---|
| I | By-pass | Tube length and UV sensor volume | Retention time and UV |
| II | Packed bed | Column void and porosity | UV |
| III | Linear gradient | and (Yamamoto method) and | , and , |
| IV | Overloading | UV |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tallvod, S.; Andersson, N.; Nilsson, B. Automation of Modeling and Calibration of Integrated Preparative Protein Chromatography Systems. Processes 2022, 10, 945. https://doi.org/10.3390/pr10050945
Tallvod S, Andersson N, Nilsson B. Automation of Modeling and Calibration of Integrated Preparative Protein Chromatography Systems. Processes. 2022; 10(5):945. https://doi.org/10.3390/pr10050945
Chicago/Turabian StyleTallvod, Simon, Niklas Andersson, and Bernt Nilsson. 2022. "Automation of Modeling and Calibration of Integrated Preparative Protein Chromatography Systems" Processes 10, no. 5: 945. https://doi.org/10.3390/pr10050945