
Adipose Tissue Immunomodulation and Treg/Th17 Imbalance in the Impaired Glucose Metabolism of Children with Obesity

 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
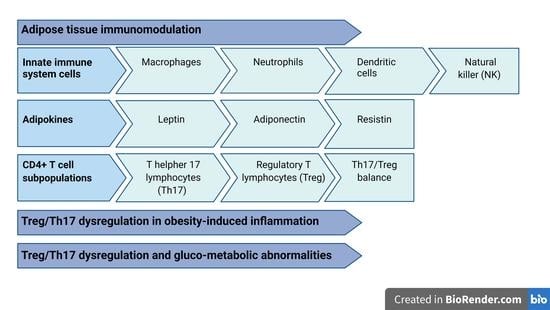
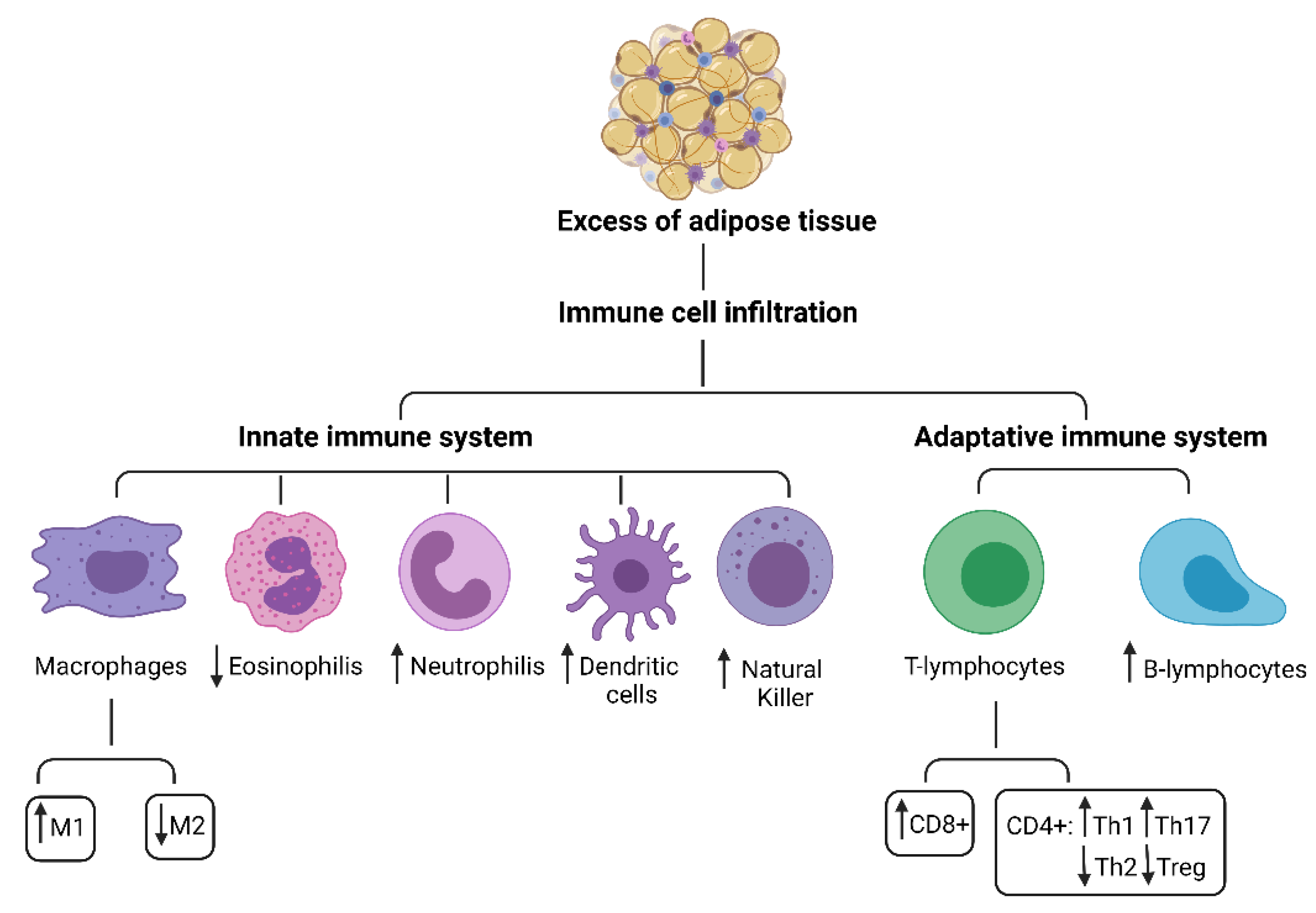
3. Adipose Tissue Immunomodulation
3.1. Innate Immune System Cells in Adipose Tissue
3.2. Adipokine Immunological Properties
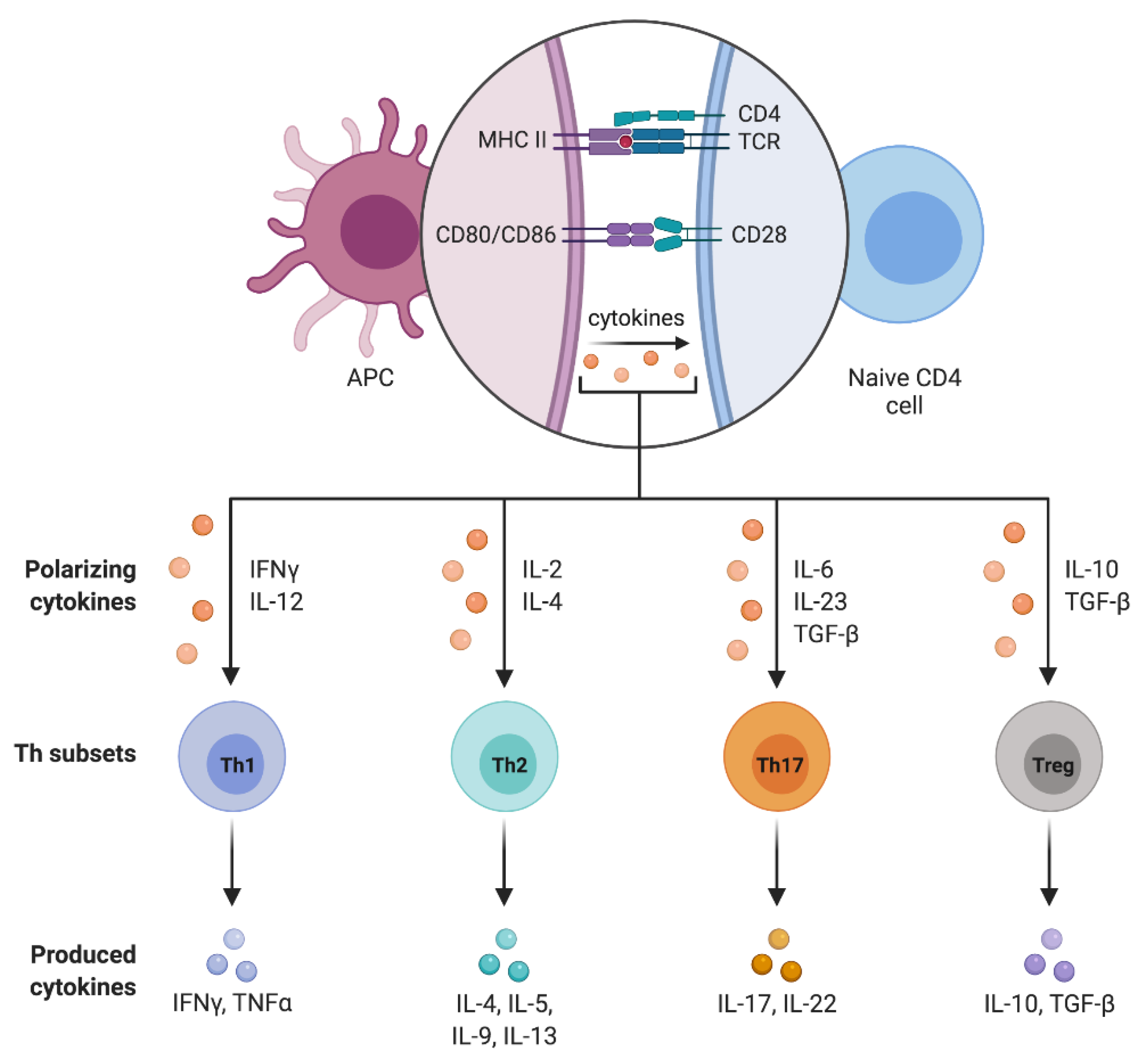
4. CD4+ T Cell Subpopulations
4.1. T Helpher 17 Lymphocytes (Th17)
4.2. Regulatory T Lymphocytes (Treg)
4.3. Treg/Th17 Balance
5. Th17 and Treg Dysregulation in Obesity-Induced Inflammation
6. Treg/Th17 Dysregulation and Gluco-Metabolic Abnormalities
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Henao-Mejia, J.; Henrickson, S.E. Obesity and immune status in children. Curr. Opin. Pediatr. 2020, 32, 805–815. [Google Scholar] [CrossRef]
- World Health Organization. WHO. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. (accessed on 18 May 2021).
- Kumar, S.; Kelly, A.S. Review of childhood obesity: From epidemiology, etiology, and comorbidities to clinical assessment and treatment. Mayo Clin. Proc. 2017, 92, 251–265. [Google Scholar] [CrossRef] [Green Version]
- Umer, A.; Kelley, G.A.; Cottrell, L.E.; Giacobbi, P.; Innes, K.E.; Lilly, C.L. Childhood obesity and adult cardiovascular disease risk factors: A systematic review with meta-analysis. BMC Public Health 2017, 17, 683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nehus, E.; Mitsnefes, M. Childhood obesity and the metabolic syndrome. Pediatr. Clin. N. Am. 2019, 66, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Childs, C.E.; Calder, P.C.; Miles, E.A. Diet and immune function. Nutrients 2019, 11, 1933. [Google Scholar] [CrossRef] [Green Version]
- Lynch, L.; Nowak, M.; Varghese, B.; Clark, J.; Hogan, A.E.; Toxavidis, V.; Balk, S.P.; O’Shea, D.; O’Farrelly, C.; Exley, M.A. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity 2012, 37, 574–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Liu, F. Regulation, communication, and functional roles of adipose tissue-resident CD4+ T cells in the control of metabolic homeostasis. Front. Immunol. 2018, 9, 1961. [Google Scholar] [CrossRef]
- Yoshida, S.; Haque, A.; Mizobuchi, T.; Iwata, T.; Chiyo, M.; Webb, T.J.; Baldridge, L.A.; Heidler, K.M.; Cummings, O.W.; Fujisawa, T.; et al. Anti-Type V collagen lymphocytes that express IL-17 and IL-23 induce rejection pathology in fresh and well-healed lung transplants. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2006, 6, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Burrell, B.E.; Bishop, D.K. Th17 cells and transplant acceptance. Transplantation 2010, 90, 945–948. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S. N aturally A rising CD4 + R egulatory T C ells for I mmunologic S elf -T olerance and N egative C ontrol of I mmune R esponses. Annu. Rev. Immunol. 2004, 22, 531–562. [Google Scholar] [CrossRef]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming growth factor-β controls development, homeostasis, and tolerance of T cells by regulatory t cell-dependent and -independent mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaur, P.; Qadir, G.A.; Upadhyay, S.; Singh, A.K.; Shukla, N.K.; Das, S.N. Skewed immunological balance between Th17 (CD4+IL17A+) and Treg (CD4+CD25+FOXP3+) cells in human oral squamous cell carcinoma. Cell. Oncol. 2012, 35, 335–343. [Google Scholar] [CrossRef]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.-H.; Pagès, F.; et al. Clinical impact of different classes of infiltrating t cytotoxic and helper cells (Th1, Th2, Treg, Th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcaterra, V.; Croce, S.; Vinci, F.; De Silvestri, A.; Cordaro, E.; Regalbuto, C.; Zuccotti, G.V.; Mameli, C.; Albertini, R.; Avanzini, M.A. Th17 and Treg balance in children with obesity and metabolically altered status. Front. Pediatr. 2020, 8, 591012. [Google Scholar] [CrossRef]
- Wen, J.; Liu, Q.; Liu, M.; Wang, B.; Li, M.; Wang, M.; Shi, X.; Liu, H.; Wu, J. Increasing imbalance of Treg/Th17 indicates more severe glucose metabolism dysfunction in overweight/obese patients. Arch. Med. Res. 2021, 52, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Richard, A.J.; White, U.; Elks, C.M.; Stephens, J.M. Adipose tissue: Physiology to metabolic dysfunction. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Carobbio, S.; Pellegrinelli, V.; Vidal-Puig, A. Adipose tissue function and expandability as determinants of lipotoxicity and the metabolic syndrome. In Obesity and Lipotoxicity; Advances in Experimental Medicine and Biology; Engin, A.B., Engin, A., Eds.; Springer International Publishing: Cham, Switzerland, 2017; Volume 960, pp. 161–196. ISBN 978-3-319-48380-1. [Google Scholar]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.-H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Au-Yong, I.T.H.; Thorn, N.; Ganatra, R.; Perkins, A.C.; Symonds, M.E. Brown adipose tissue and seasonal variation in humans. Diabetes 2009, 58, 2583–2587. [Google Scholar] [CrossRef] [Green Version]
- Hibi, M.; Oishi, S.; Matsushita, M.; Yoneshiro, T.; Yamaguchi, T.; Usui, C.; Yasunaga, K.; Katsuragi, Y.; Kubota, K.; Tanaka, S.; et al. Brown adipose tissue is involved in diet-induced thermogenesis and whole-body fat utilization in healthy humans. Int. J. Obes. 2016, 40, 1655–1661. [Google Scholar] [CrossRef] [Green Version]
- Cinti, S. The Adipose Organ at a Glance. Dis. Model. Mech. 2012, 5, 588–594. [Google Scholar] [CrossRef] [Green Version]
- Hausman, D.B.; DiGirolamo, M.; Bartness, T.J.; Hausman, G.J.; Martin, R.J. The biology of white adipocyte proliferation. Obes. Rev. 2001, 2, 239–254. [Google Scholar] [CrossRef]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Hosoya, Y.; Yamashita, H.; Fujita, H.; Ohsugi, M.; Tobe, K.; Kadowaki, T.; Nagai, R.; et al. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes 2007, 56, 1517–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galic, S.; Oakhill, J.S.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef]
- Kelishadi, R.; Roufarshbaf, M.; Soheili, S.; Payghambarzadeh, F.; Masjedi, M. Association of childhood obesity and the immune system: A systematic review of reviews. Child. Obes. 2017, 13, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Frayn, K.N.; Karpe, F.; Fielding, B.A.; Macdonald, I.A.; Coppack, S.W. Integrative physiology of human adipose tissue. Int. J. Obes. 2003, 27, 875–888. [Google Scholar] [CrossRef] [Green Version]
- Calcaterra, V.; Regalbuto, C.; Porri, D.; Pelizzo, G.; Mazzon, E.; Vinci, F.; Zuccotti, G.; Fabiano, V.; Cena, H. Inflammation in obesity-related complications in children: The protective effect of diet and its potential role as a therapeutic agent. Biomolecules 2020, 10, 1324. [Google Scholar] [CrossRef]
- Maurya, R.; Bhattacharya, P.; Dey, R.; Nakhasi, H.L. Leptin functions in infectious diseases. Front. Immunol. 2018, 9, 2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Inflammatory pathways and insulin action. Int. J. Obes. 2003, 27, S53–S55. [Google Scholar] [CrossRef] [Green Version]
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 2007, 56, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Rausch, M.E.; Weisberg, S.; Vardhana, P.; Tortoriello, D.V. Obesity in C57BL/6J Mice is characterized by adipose tissue hypoxia and cytotoxic T-Cell infiltration. Int. J. Obes. 2008, 32, 451–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Gao, Z.; Yin, J.; He, Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of Ob/Ob and dietary obese mice. Am. J. Physiol.-Endocrinol. Metab. 2007, 293, E1118–E1128. [Google Scholar] [CrossRef] [Green Version]
- Ye, J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int. J. Obes. 2009, 33, 54–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahimi-Horn, M.C.; Pouysségur, J. Oxygen, A source of life and stress. FEBS Lett. 2007, 581, 3582–3591. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- McArdle, M.A.; Finucane, O.M.; Connaughton, R.M.; McMorrow, A.M.; Roche, H.M. Mechanisms of obesity-induced inflammation and insulin resistance: Insights into the emerging role of nutritional strategies. Front. Endocrinol. 2013, 4, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Ma, Y.; Cui, Q.; Xu, J.; Tang, Z.; Wang, Y.; He, C.; Wang, X. Toll-like receptor 4 plays a key role in advanced glycation end products-induced M1 macrophage polarization. Biochem. Biophys. Res. Commun. 2020, 531, 602–608. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Investig. 2003, 112, 1785–1788. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling: Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Pangrazzi, L.; Naismith, E.; Miggitsch, C.; Carmona Arana, J.A.; Keller, M.; Grubeck-Loebenstein, B.; Weinberger, B. The impact of body mass index on adaptive immune cells in the human bone marrow. Immun. Ageing 2020, 17, 15. [Google Scholar] [CrossRef]
- Alwarawrah, Y.; Kiernan, K.; MacIver, N.J. Changes in nutritional status impact immune cell metabolism and function. Front. Immunol. 2018, 9, 1055. [Google Scholar] [CrossRef] [Green Version]
- Zhuge, F.; Ni, Y.; Nagashimada, M.; Nagata, N.; Xu, L.; Mukaida, N.; Kaneko, S.; Ota, T. DPP-4 inhibition by linagliptin attenuates obesity-related inflammation and insulin resistance by regulating M1/M2 macrophage polarization. Diabetes 2016, 65, 2966–2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, C.A.; Lingaraju, A.; Schoenfelt, K.Q.; Zhou, G.; Cui, C.; Jacobs-El, H.; Babenko, I.; Hoofnagle, A.; Czyz, D.; Shuman, H.; et al. Obesity and insulin resistance promote atherosclerosis through an IFNγ-regulated macrophage protein network. Cell Rep. 2018, 23, 3021–3030. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose tissue macrophage-derived exosomal MiRNAs can modulate in vivo and in vitro insulin sensitivity. Cell 2017, 171, 372–384.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brotfain, E.; Hadad, N.; Shapira, Y.; Avinoah, E.; Zlotnik, A.; Raichel, L.; Levy, R. Neutrophil functions in morbidly obese subjects. Clin. Exp. Immunol. 2015, 181, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- D’Abbondanza, M.; Martorelli, E.E.; Ricci, M.A.; De Vuono, S.; Migliola, E.N.; Godino, C.; Corradetti, S.; Siepi, D.; Paganelli, M.T.; Maugeri, N.; et al. Increased Plasmatic NETs by-products in patients in severe obesity. Sci. Rep. 2019, 9, 14678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, C.; Wadowski, M.; Goruk, S.; Cameron, L.; Sharma, A.M.; Field, C.J. Individuals with obesity and type 2 diabetes have additional immune dysfunction compared with obese individuals who are metabolically healthy. BMJ Open Diabetes Res. Care 2017, 5, e000379. [Google Scholar] [CrossRef]
- O’Shea, D.; Corrigan, M.; Dunne, M.R.; Jackson, R.; Woods, C.; Gaoatswe, G.; Moynagh, P.N.; O’Connell, J.; Hogan, A.E. Changes in human dendritic cell number and function in severe obesity may contribute to increased susceptibility to viral infection. Int. J. Obes. 2013, 37, 1510–1513. [Google Scholar] [CrossRef] [Green Version]
- Mattioli, B.; Straface, E.; Quaranta, M.G.; Giordani, L.; Viora, M. Leptin promotes differentiation and survival of human dendritic cells and licenses them for Th1 priming. J. Immunol. 2005, 174, 6820–6828. [Google Scholar] [CrossRef] [PubMed]
- Bertola, A.; Ciucci, T.; Rousseau, D.; Bourlier, V.; Duffaut, C.; Bonnafous, S.; Blin-Wakkach, C.; Anty, R.; Iannelli, A.; Gugenheim, J.; et al. Identification of Adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 2012, 61, 2238–2247. [Google Scholar] [CrossRef] [Green Version]
- Moraes-Vieira, P.M.M.; Larocca, R.A.; Bassi, E.J.; Peron, J.P.S.; Andrade-Oliveira, V.; Wasinski, F.; Araujo, R.; Thornley, T.; Quintana, F.J.; Basso, A.S.; et al. Leptin deficiency impairs maturation of dendritic cells and enhances induction of regulatory T and Th17 cells: Immunomodulation. Eur. J. Immunol. 2014, 44, 794–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-H.; Jung, H.-J.; Kim, T.S. IL-33 changes CD25hi tregs to Th17 cells through a dendritic cell-mediated pathway. Immunol. Lett. 2020, 218, 5–10. [Google Scholar] [CrossRef]
- O’Sullivan, T.E.; Sun, J.C.; Lanier, L.L. Natural killer cell memory. Immunity 2015, 43, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Bähr, I.; Spielmann, J.; Quandt, D.; Kielstein, H. Obesity-associated alterations of natural killer cells and immunosurveillance of cancer. Front. Immunol. 2020, 11, 245. [Google Scholar] [CrossRef] [Green Version]
- Viel, S.; Besson, L.; Charrier, E.; Marçais, A.; Disse, E.; Bienvenu, J.; Walzer, T.; Dumontet, C. Alteration of natural killer cell phenotype and function in obese individuals. Clin. Immunol. 2017, 177, 12–17. [Google Scholar] [CrossRef]
- Wouters, K.; Gaens, K.; Bijnen, M.; Verboven, K.; Jocken, J.; Wetzels, S.; Wijnands, E.; Hansen, D.; Van Greevenbroek, M.; Duijvestijn, A.; et al. Circulating classical monocytes are associated with CD11C+ macrophages in human visceral adipose tissue. Sci. Rep. 2017, 7, 42665. [Google Scholar] [CrossRef]
- Guo, H.; Xu, B.; Gao, L.; Sun, X.; Qu, X.; Li, X.; Liu, S.; Feng, J.; Wang, J.; Tang, Y.; et al. High frequency of activated natural killer and natural killer T-cells in patients with new onset of type 2 diabetes mellitus. Exp. Biol. Med. 2012, 237, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Wensveen, F.M.; Jelenčić, V.; Valentić, S.; Šestan, M.; Wensveen, T.T.; Theurich, S.; Glasner, A.; Mendrila, D.; Štimac, D.; Wunderlich, F.T.; et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat. Immunol. 2015, 16, 376–385. [Google Scholar] [CrossRef]
- Lee, B.-C.; Kim, M.-S.; Pae, M.; Yamamoto, Y.; Eberlé, D.; Shimada, T.; Kamei, N.; Park, H.-S.; Sasorith, S.; Woo, J.R.; et al. Adipose natural killer cells regulate adipose tissue macrophages to promote insulin resistance in obesity. Cell Metab. 2016, 23, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Perez, C.; Fernandez-Galaz, C.; Fernandez-Agullo, T.; Arribas, C.; Andres, A.; Ros, M.; Carrascosa, J.M. Leptin impairs insulin signaling in rat adipocytes. Diabetes 2004, 53, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceddia, R.; William, W.N.; Lima, F.; Curi, R. Leptin inhibits insulin-stimulated incorporation of glucose into lipids and stimulates glucose decarboxylation in isolated rat adipocytes. J. Endocrinol. 1998, 158, R7–R9. [Google Scholar] [CrossRef]
- Kumar, R.; Mal, K.; Razaq, M.K.; Magsi, M.; Memon, M.K.; Memon, S.; Afroz, M.N.; Siddiqui, H.F.; Rizwan, A. Association of leptin with obesity and insulin resistance. Cureus 2020, 12, e12178. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Flier, J.S. Leptin. Annu. Rev. Physiol. 2000, 62, 413–437. [Google Scholar] [CrossRef] [Green Version]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, fat mass and immune system: Role for leptin. Front. Physiol. 2018, 9, 640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Cava, A. Leptin in inflammation and autoimmunity. Cytokine 2017, 98, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.V.; Scherer, P.E. Adiponectin, the past two decades. J. Mol. Cell Biol. 2016, 8, 93–100. [Google Scholar] [CrossRef]
- Ohashi, K.; Shibata, R.; Murohara, T.; Ouchi, N. Role of anti-inflammatory adipokines in obesity-related diseases. Trends Endocrinol. Metab. 2014, 25, 348–355. [Google Scholar] [CrossRef]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Weyer, C.; Funahashi, T.; Tanaka, S.; Hotta, K.; Matsuzawa, Y.; Pratley, R.E.; Tataranni, P.A. Hypoadiponectinemia in obesity and type 2 diabetes: Close association with insulin resistance and hyperinsulinemia. J. Clin. Endocrinol. Metab. 2001, 86, 1930–1935. [Google Scholar] [CrossRef]
- Cnop, M.; Havel, P.J.; Utzschneider, K.M.; Carr, D.B.; Sinha, M.K.; Boyko, E.J.; Retzlaff, B.M.; Knopp, R.H.; Brunzell, J.D.; Kahn, S.E. Relationship of adiponectin to body fat distribution, insulin sensitivity and plasma lipoproteins: Evidence for independent roles of age and sex. Diabetologia 2003, 46, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Côté, M.; Mauriège, P.; Bergeron, J.; Alméras, N.; Tremblay, A.; Lemieux, I.; Després, J.-P. Adiponectinemia in visceral obesity: Impact on glucose tolerance and plasma lipoprotein and lipid levels in men. J. Clin. Endocrinol. Metab. 2005, 90, 1434–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef]
- Silswal, N.; Singh, A.K.; Aruna, B.; Mukhopadhyay, S.; Ghosh, S.; Ehtesham, N.Z. Human resistin stimulates the pro-inflammatory cytokines TNF-α and IL-12 in macrophages by NF-ΚB-Dependent pathway. Biochem. Biophys. Res. Commun. 2005, 334, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Bokarewa, M.; Nagaev, I.; Dahlberg, L.; Smith, U.; Tarkowski, A. Resistin, an adipokine with potent proinflammatory properties. J. Immunol. 2005, 174, 5789–5795. [Google Scholar] [CrossRef]
- Filková, M.; Haluzík, M.; Gay, S.; Šenolt, L. The role of resistin as a regulator of inflammation: Implications for various human pathologies. Clin. Immunol. 2009, 133, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamane, H.; Paul, W.E. Early signaling events that underlie fate decisions of naive CD4 + T cells toward distinct t-helper cell subsets. Immunol. Rev. 2013, 252, 12–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrone, T.; Jirillo, E. Childhood obesity: Immune response and nutritional approaches. Front. Immunol. 2015, 6, 76. [Google Scholar] [CrossRef] [Green Version]
- Ryba-Stanisławowska, M.; Skrzypkowska, M.; Myśliwiec, M.; Myśliwska, J. Loss of the balance between CD4+Foxp3+ Regulatory T cells and CD4+IL17A+ Th17 cells in patients with type 1 diabetes. Hum. Immunol. 2013, 74, 701–707. [Google Scholar] [CrossRef]
- Cipolletta, D. Adipose tissue-resident regulatory T cells: Phenotypic specialization, functions and therapeutic potential. Immunology 2014, 142, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Kälin, S.; Becker, M.; Ott, V.B.; Serr, I.; Hosp, F.; Mollah, M.M.H.; Keipert, S.; Lamp, D.; Rohner-Jeanrenaud, F.; Flynn, V.K.; et al. A Stat6/Pten axis links regulatory T cells with adipose tissue function. Cell Metab. 2017, 26, 475–492.e7. [Google Scholar] [CrossRef] [Green Version]
- Gyllenhammer, L.E.; Lam, J.; Alderete, T.L.; Allayee, H.; Akbari, O.; Katkhouda, N.; Goran, M.I. Lower omental T-regulatory cell count is associated with higher fasting glucose and lower β-cell function in adults with obesity: Lower omental tregs and type 2 diabetes risk. Obesity 2016, 24, 1274–1282. [Google Scholar] [CrossRef] [Green Version]
- Yuan, N.; Zhang, H.; Wei, Q.; Wang, P.; Guo, W. Expression of CD4+CD25+Foxp3+ regulatory T cells, interleukin 10 and transforming growth factor β in newly diagnosed type 2 diabetic patients. Exp. Clin. Endocrinol. Diabetes 2018, 126, 96–101. [Google Scholar] [CrossRef]
- Wang, M.; Chen, F.; Wang, J.; Zeng, Z.; Yang, Q.; Shao, S. Th17 and Treg lymphocytes in obesity and type 2 diabetic patients. Clin. Immunol. 2018, 197, 77–85. [Google Scholar] [CrossRef]
- Gutcher, I.; Donkor, M.K.; Ma, Q.; Rudensky, A.Y.; Flavell, R.A.; Li, M.O. Autocrine transforming growth factor-Β1 promotes in vivo Th17 cell differentiation. Immunity 2011, 34, 396–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, K.; Kishimoto, T. Th17 Cells: Inflammation and regulation. Atlas Genet. Cytogenet. Oncol. Haematol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Brucklacher-Waldert, V.; Steinbach, K.; Lioznov, M.; Kolster, M.; Hölscher, C.; Tolosa, E. Phenotypical characterization of human Th17 cells unambiguously identified by surface IL-17A expression. J. Immunol. 2009, 183, 5494–5501. [Google Scholar] [CrossRef]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The Orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Ghoreschi, K.; Laurence, A.; Yang, X.-P.; Tato, C.M.; McGeachy, M.J.; Konkel, J.E.; Ramos, H.L.; Wei, L.; Davidson, T.S.; Bouladoux, N.; et al. Generation of pathogenic TH17 cells in the absence of TGF-β signalling. Nature 2010, 467, 967–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarantino, G.; Costantini, S.; Finelli, C.; Capone, F.; Guerriero, E.; La Sala, N.; Gioia, S.; Castello, G. Is serum Interleukin-17 associated with early atherosclerosis in obese patients? J. Transl. Med. 2014, 12, 214. [Google Scholar] [CrossRef] [Green Version]
- Mottaghi, A.; Ebrahimof, S.; Angoorani, P.; Saboor-Yaraghi, A.-A. Vitamin a supplementation reduces IL-17 and RORc gene expression in atherosclerotic patients. Scand. J. Immunol. 2014, 80, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matarese, G.; Procaccini, C.; De Rosa, V.; Horvath, T.L.; La Cava, A. Regulatory T cells in obesity: The leptin connection. Trends Mol. Med. 2010, 16, 247–256. [Google Scholar] [CrossRef]
- O’Garra, A.; Vieira, P. Regulatory T cells and mechanisms of immune system control. Nat. Med. 2004, 10, 801–805. [Google Scholar] [CrossRef]
- Taylor, S.R.J.; Alexander, D.R.; Cooper, J.C.; Higgins, C.F.; Elliott, J.I. Regulatory T cells are resistant to apoptosis via TCR but Not P2X 7. J. Immunol. 2007, 178, 3474–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, S.J.; Rajotte, R.V.; Korbutt, G.S.; Bleackley, R.C. Granzyme B: A natural born killer. Immunol. Rev. 2003, 193, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Liu, M.; Cui, M.; Li, T. Granzyme B-expressing Treg cells are enriched in colorectal cancer and present the potential to eliminate autologous T conventional cells. Immunol. Lett. 2020, 217, 7–14. [Google Scholar] [CrossRef]
- Campbell, C.; Rudensky, A. Roles of regulatory T cells in tissue pathophysiology and metabolism. Cell Metab. 2020, 31, 18–25. [Google Scholar] [CrossRef]
- Qiu, H.; He, Y.; Ouyang, F.; Jiang, P.; Guo, S.; Guo, Y. The role of regulatory T cells in pulmonary arterial hypertension. J. Am. Heart Assoc. 2019, 8, e014201. [Google Scholar] [CrossRef]
- Jin, W.; Cui, B.; Li, P.; Hua, F.; Lv, X.; Zhou, J.; Hu, Z.; Zhang, X. 1,25-Dihydroxyvitamin D3 protects obese rats from metabolic syndrome via promoting regulatory T cell-mediated resolution of inflammation. Acta Pharm. Sin. B 2018, 8, 178–187. [Google Scholar] [CrossRef]
- Zhong, J.; Rao, X.; Braunstein, Z.; Taylor, A.; Narula, V.; Hazey, J.; Mikami, D.; Needleman, B.; Rutsky, J.; Sun, Q.; et al. T-cell costimulation protects obesity-induced adipose inflammation and insulin resistance. Diabetes 2014, 63, 1289–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, H.; Li, R.; Hu, H.; Hu, Y.; Chen, X. Modulation of regulatory T cell activity by TNF receptor type II-targeting pharmacological agents. Front. Immunol. 2018, 9, 594. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Gaffen, S.L. IL-17 in obesity and adipogenesis. Cytokine Growth Factor Rev. 2010, 21, 449–453. [Google Scholar] [CrossRef] [Green Version]
- Lapierre, P.; Béland, K.; Yang, R.; Alvarez, F. Adoptive transfer of Ex Vivo expanded regulatory T cells in an autoimmune hepatitis murine model restores peripheral tolerance. Hepatology 2013, 57, 217–227. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Lian, Z.-X.; Moritoki, Y.; Lan, R.Y.; Tsuneyama, K.; Chuang, Y.-H.; Yang, G.-X.; Ridgway, W.; Ueno, Y.; Ansari, A.A.; et al. IL-2 receptor A −/− mice and the development of primary biliary cirrhosis. Hepatology 2006, 44, 1240–1249. [Google Scholar] [CrossRef]
- Kikuchi, J.; Hashizume, M.; Kaneko, Y.; Yoshimoto, K.; Nishina, N.; Takeuchi, T. Peripheral blood CD4+CD25+CD127low regulatory T cells are significantly increased by tocilizumab treatment in patients with rheumatoid arthritis: Increase in regulatory T cells correlates with clinical response. Arthritis Res. Ther. 2015, 17, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neurath, M.F.; Weigmann, B.; Finotto, S.; Glickman, J.; Nieuwenhuis, E.; Iijima, H.; Mizoguchi, A.; Mizoguchi, E.; Mudter, J.; Galle, P.R.; et al. The transcription factor T-Bet regulates mucosal t cell activation in experimental colitis and crohn’s disease. J. Exp. Med. 2002, 195, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Gaublomme, J.T.; Yosef, N.; Lee, Y.; Gertner, R.S.; Yang, L.V.; Wu, C.; Pandolfi, P.P.; Mak, T.; Satija, R.; Shalek, A.K.; et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 2015, 163, 1400–1412. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Liu, D.; Shen, R.; Li, D.; Hu, Q.; Yan, Y.; Sun, J.; Zhang, F.; Wan, H.; Dong, P.; et al. Discovery of a novel RORγ antagonist with skin-restricted exposure for topical treatment of mild to moderate psoriasis. Sci. Rep. 2021, 11, 9132. [Google Scholar] [CrossRef] [PubMed]
- Fabrizi, M.; Marchetti, V.; Mavilio, M.; Marino, A.; Casagrande, V.; Cavalera, M.; Moreno-Navarrete, J.M.; Mezza, T.; Sorice, G.P.; Fiorentino, L.; et al. IL-21 Is a major negative regulator of IRF4-dependent lipolysis affecting tregs in adipose tissue and systemic insulin sensitivity. Diabetes 2014, 63, 2086–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, T.; Liu, L.-F.; Lamendola, C.; Shen, L.; Morton, J.; Rivas, H.; Winer, D.; Tolentino, L.; Choi, O.; Zhang, H.; et al. T-cell profile in adipose tissue is associated with insulin resistance and systemic inflammation in humans. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2637–2643. [Google Scholar] [CrossRef] [Green Version]
- Winer, S.; Chan, Y.; Paltser, G.; Truong, D.; Tsui, H.; Bahrami, J.; Dorfman, R.; Wang, Y.; Zielenski, J.; Mastronardi, F.; et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 2009, 15, 921–929. [Google Scholar] [CrossRef]
- Hu, W.; Wang, R.; Li, J.; Zhang, J.; Wang, W. Association of irisin concentrations with the presence of diabetic nephropathy and retinopathy. Ann. Clin. Biochem. Int. J. Lab. Med. 2016, 53, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Yu, H.; Sun, G.; Sun, X.; Jin, H.; Zhang, C.; Shi, W.; Tian, D.; Liu, K.; Xu, H.; et al. OX40 Promotes obesity-induced adipose inflammation and insulin resistance. Cell. Mol. Life Sci. 2017, 74, 3827–3840. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, Q.; Liu, S.; Lambrechts, M.; Qu, Y.; You, Z. AZD5363 Inhibits inflammatory synergy between interleukin-17 and insulin/insulin-like growth factor 1. Front. Oncol. 2014, 4, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Sanchez, M.E.; Hiriart, M.; Alvarez-Buylla, E.R. The CD4+ T cell regulatory network mediates inflammatory responses during acute hyperinsulinemia: A simulation study. BMC Syst. Biol. 2017, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, H.; Wang, F.; Han, L.; Liu, M.; Li, Y.; Wang, Z.; Tang, M.; Zhang, W.; Zhong, M. Overexpression of PTPN2 in visceral adipose tissue ameliorated atherosclerosis via T cells polarization shift in diabetic Apoe−/− Mice. Cell. Physiol. Biochem. 2018, 46, 118–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhun, J.; Woo, J.S.; Lee, S.H.; Jeong, J.-H.; Jung, K.; Hur, W.; Lee, S.-Y.; Ryu, J.Y.; Moon, Y.-M.; Jung, Y.J.; et al. GRIM19 impedes obesity by regulating inflammatory white fat browning and promoting Th17/Treg balance. Cells 2021, 10, 162. [Google Scholar] [CrossRef]
- Kroemer, G.; Zitvogel, L. CD4+ T Cells at the Center of Inflammaging. Cell Metab. 2020, 32, 4–5. [Google Scholar] [CrossRef]
- Nicholas, D.A.; Proctor, E.A.; Agrawal, M.; Belkina, A.C.; Van Nostrand, S.C.; Panneerseelan-Bharath, L.; Jones, A.R.; Raval, F.; Ip, B.C.; Zhu, M.; et al. Fatty acid metabolites combine with reduced β oxidation to activate Th17 inflammation in human type 2 diabetes. Cell Metab. 2019, 30, 447–461.e5. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Nikolajczyk, B.S. Tissue immune cells fuel obesity-associated inflammation in adipose tissue and beyond. Front. Immunol. 2019, 10, 1587. [Google Scholar] [CrossRef] [Green Version]
- Capone, A.; Volpe, E. Transcriptional regulators of t helper 17 cell differentiation in health and autoimmune diseases. Front. Immunol. 2020, 11, 348. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Takeuchi, Y.; Hirota, K. The pathogenicity of Th17 cells in autoimmune diseases. Semin. Immunopathol. 2019, 41, 283–297. [Google Scholar] [CrossRef]
- Pacifico, L.; Di Renzo, L.; Anania, C.; Osborn, J.F.; Ippoliti, F.; Schiavo, E.; Chiesa, C. Increased T-helper interferon-γ-secreting cells in obese children. Eur. J. Endocrinol. 2006, 154, 691–697. [Google Scholar] [CrossRef] [Green Version]
- Touch, S.; Clément, K.; André, S. T cell populations and functions are altered in human obesity and type 2 diabetes. Curr. Diab. Rep. 2017, 17, 81. [Google Scholar] [CrossRef] [Green Version]
- Duffaut, C.; Zakaroff-Girard, A.; Bourlier, V.; Decaunes, P.; Maumus, M.; Chiotasso, P.; Sengenès, C.; Lafontan, M.; Galitzky, J.; Bouloumié, A. Interplay between human adipocytes and T lymphocytes in obesity: CCL20 as an Adipochemokine and T Lymphocytes as lipogenic modulators. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1608–1614. [Google Scholar] [CrossRef] [Green Version]
- Womack, J.; Tien, P.C.; Feldman, J.; Shin, J.H.; Fennie, K.; Anastos, K.; Cohen, M.H.; Bacon, M.C.; Minkoff, H. Obesity and immune cell counts in women. Metabolism 2007, 56, 998–1004. [Google Scholar] [CrossRef] [Green Version]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The Kinase MTOR Regulates the Differentiation of Helper T Cells through the selective activation of signaling by MTORC1 and MTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Procaccini, C.; De Rosa, V.; Galgani, M.; Carbone, F.; Cassano, S.; Greco, D.; Qian, K.; Auvinen, P.; Calì, G.; Stallone, G.; et al. Leptin-induced MTOR activation defines a specific molecular and transcriptional signature controlling CD4 + Effector T cell responses. J. Immunol. 2012, 189, 2941–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Yang, J.; Wang, X.; Li, D.; Lv, L.; Li, B. Th17 cells in autoimmune diseases. Front. Med. 2015, 9, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, J.B.; Ferraro, A.A.; Sananez, I.; Gancedo, M.C.; Baz, P.; Billordo, L.A.; Fainboim, L.; Arruvito, L. ATP-induced inflammation drives tissue-resident Th17 cells in metabolically unhealthy obesity. J. Immunol. 2016, 196, 3287–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega-Cárdenas, M.; Uresti-Rivera, E.E.; Cortés-García, J.D.; Briones-Espinoza, M.; Ruíz-Rodríguez, V.M.; Reynaga-Hernández, E.; Mendez-Mancilla, A.; Portales-Pérez, D.P. Increased levels of adipose tissue-resident Th17 cells in obesity associated with MiR-326. Immunol. Lett. 2019, 211, 60–67. [Google Scholar] [CrossRef]
- Švec, P.; Vásárhelyi, B.; Pászthy, B.; Körner, A.; Kovács, L.; Tulassay, T.; Treszl, A. Do regulatory T cells contribute to Th1 skewness in obesity? Exp. Clin. Endocrinol. Diabetes 2007, 115, 439–443. [Google Scholar] [CrossRef]
- van der Weerd, K.; Dik, W.A.; Schrijver, B.; Schweitzer, D.H.; Langerak, A.W.; Drexhage, H.A.; Kiewiet, R.M.; van Aken, M.O.; van Huisstede, A.; Van Dongen, J.J.M.; et al. Morbidly obese human subjects have increased peripheral blood CD4+ T cells with skewing toward a Treg-and Th2-dominated phenotype. Diabetes 2012, 61, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Zhi, C.; Huang, J.; Wang, J.; Cao, H.; Bai, Y.; Guo, J.; Su, Z. Connection between gut microbiome and the development of obesity. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1987–1998. [Google Scholar] [CrossRef]
- Vallianou, N.; Stratigou, T.; Christodoulatos, G.S.; Dalamaga, M. Understanding the role of the gut microbiome and microbial metabolites in obesity and obesity-associated metabolic disorders: Current evidence and perspectives. Curr. Obes. Rep. 2019, 8, 317–332. [Google Scholar] [CrossRef]
- Todosenko, N.; Vulf, M.; Yurova, K.; Skuratovskaia, D.; Khaziakhmatova, O.; Gazatova, N.; Melashchenko, O.; Urazova, O.; Litvinova, L. The pathogenic subpopulation of Th17 cells in obesity. Curr. Pharm. Des. 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, H.J.; Nagpal, R.; Yadav, H. Microbiome-immune-metabolic axis in the epidemic of childhood obesity: Evidence and opportunities. Obes Rev. 2020, 21, e12963. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Afourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature. 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Mar Teau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Parekh, P.J.; Balart, L.A.; Johnson, D.A. The influence of the gut microbiome on obesity, metabolic syndrome and gastrointestinal disease. Clin. Transl. Gastroenterol. 2015, 6, e91. [Google Scholar] [CrossRef]
- >Ou, W.; Hu, H.; Yang, P.; Dai, J.; Ai, Q.; Zhang, W. Dietary daidzein improved intestinal health of juvenile turbot in terms of intestinal mucosal barrier function and intestinal microbiota fish shellfish. Immunology 2019, 19, 132–141. [Google Scholar]
- Caruso, R.; Lo, B.C.; Núñez, G. Host–microbiota Interactions in Inflammatory Bowel Disease. Nat. Rev. Immunol. 2020, 20, 411–426. [Google Scholar] [CrossRef]
- Abo, H.; Chassaing, B.; Harusato, A.; Quiros, M.; Brazil, J.C.; Ngo, V.L. Erythroid differentiation regulator-1 induced by microbiota in early life drives intestinal stem cell proliferation and regeneration. Nat. Commun. 2020. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- <named-content content-type="background:white">Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Tang, D.; Wang, Y.; Kang, W.; Jiannan, Z.; Dong, R.; Feng, Q. Chitosan attenuates obesity by modifying the intestinal microbiota and increasing serum leptin levels in mice. J. Funct. Foods. 2020, 64, 103659. [Google Scholar] [CrossRef]
- Santos-Marcos, A.; Perez-Jimenez, F.; Camargo, A. The role of diet and intestinal microbiota in the development of metabolic syndrome. J. Nutr. Biochem. 2019, 70, 1–27. [Google Scholar] [CrossRef]
- Luck, H.; Khan, S.; Kim, J.H.; Copeland, J.K.; Revelo, X.S.; Tsai, S. Gut-associated IgA+ immune cells regulate obesity-related insulin resistance. Nat. Commun. 2019, 10, 3650. [Google Scholar] [CrossRef]
- Sun, L.; Fu, J.; Zhou, Y. Metabolism controls the balance of Th17/T-regulatory cells. Front. Immunol. 2017, 8, 1632. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Lopes, J.E.; Chong, M.M.W.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, T.J.; Grosso, J.F.; Yen, H.-R.; Xin, H.; Kortylewski, M.; Albesiano, E.; Hipkiss, E.L.; Getnet, D.; Goldberg, M.V.; Maris, C.H.; et al. Cutting edge: An in vivo requirement for STAT3 signaling in T H 17 development and T H 17-dependent autoimmunity. J. Immunol. 2007, 179, 4313–4317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becher, B.; Durell, B.G.; Noelle, R.J. IL-23 Produced by CNS-resident cells controls T cell encephalitogenicity during the effector phase of experimental autoimmune encephalomyelitis. J. Clin. Investig. 2003, 112, 1186–1191. [Google Scholar] [CrossRef] [Green Version]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-β induces development of the TH17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L. Metabolism in T cell activation and differentiation. Curr. Opin. Immunol. 2010, 22, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Green, D.R. The immune diet: Meeting the metabolic demands of lymphocyte activation. F1000 Biol. Rep. 2012, 4, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Green, D.R. Metabolic reprogramming and metabolic dependency in T cells. Immunol. Rev. 2012, 249, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerriets, V.A.; Rathmell, J.C. Metabolic pathways in T cell fate and function. Trends Immunol. 2012, 33, 168–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, R.; Junttila, I.S.; Wei, G.; Urban, J.F.; Zhao, K.; Paul, W.E.; Zhu, J. The transcription factor GATA3 actively represses RUNX3 protein-regulated production of interferon-γ. Immunity 2010, 32, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Deiuliis, J.; Shah, Z.; Shah, N.; Needleman, B.; Mikami, D.; Narula, V.; Perry, K.; Hazey, J.; Kampfrath, T.; Kollengode, M.; et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS ONE 2011, 6, e16376. [Google Scholar] [CrossRef] [Green Version]
- Wagner, N.-M.; Brandhorst, G.; Czepluch, F.; Lankeit, M.; Eberle, C.; Herzberg, S.; Faustin, V.; Riggert, J.; Oellerich, M.; Hasenfuss, G.; et al. Circulating regulatory T cells are reduced in obesity and may identify subjects at increased metabolic and cardiovascular risk: Regulatory T cells and obesity. Obesity 2013, 21, 461–468. [Google Scholar] [CrossRef]
- Eller, K.; Kirsch, A.; Wolf, A.M.; Sopper, S.; Tagwerker, A.; Stanzl, U.; Wolf, D.; Patsch, W.; Rosenkranz, A.R.; Eller, P. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes 2011, 60, 2954–2962. [Google Scholar] [CrossRef] [Green Version]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. PPAR-γ Is a major driver of the accumulation and phenotype of adipose tissue treg cells. Nature 2012, 486, 549–553. [Google Scholar] [CrossRef]
- Tao, L.; Liu, H.; Gong, Y. Role and mechanism of the Th17/Treg cell balance in the development and progression of insulin resistance. Mol. Cell. Biochem. 2019, 459, 183–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilleron, J.; Bouget, G.; Ivanov, S.; Meziat, C.; Ceppo, F.; Vergoni, B.; Djedaini, M.; Soprani, A.; Dumas, K.; Jacquel, A.; et al. Rab4b deficiency in T cells promotes adipose Treg/Th17 imbalance, adipose tissue dysfunction, and insulin resistance. Cell Rep. 2018, 25, 3329–3341.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalska, I.; Straczkowski, M.; Nikolajuk, A.; Adamska, A.; Karczewska-Kupczewska, M.; Otziomek, E.; Kinalska, I.; Gorska, M. Insulin resistance, serum adiponectin, and proinflammatory markers in young subjects with the metabolic syndrome. Metabolism 2008, 57, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Inflammatory mechanisms in the regulation of insulin resistance. Mol. Med. 2008, 14, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, L.; Rőszer, T.; Ricote, M. Inflammatory mediators and insulin resistance in obesity: Role of nuclear receptor signaling in macrophages. Mediators Inflamm. 2010, 2010, 1–10. [Google Scholar] [CrossRef]
- Han, J.M.; Patterson, S.J.; Speck, M.; Ehses, J.A.; Levings, M.K. Insulin inhibits IL-10–mediated regulatory T cell function: Implications for obesity. J. Immunol. 2014, 192, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Sumarac-Dumanovic, M.; Jeremic, D.; Pantovic, A.; Janjetovic, K.; Stamenkovic-Pejkovic, D.; Cvijovic, G.; Stevanovic, D.; Micic, D.; Trajkovic, V. Therapeutic improvement of glucoregulation in newly diagnosed type 2 diabetes patients is associated with a reduction of IL-17 levels. Immunobiology 2013, 218, 1113–1118. [Google Scholar] [CrossRef]
- Amoani, B.; Sakyi, S.A.; Mantey, R.; Laing, E.F.; Ephraim, R.D.; Sarfo-Katanka, O.; Koffie, S.; Obese, E.; Afranie, B.O. Increased metformin dosage suppresses pro-inflammatory cytokine levels in systemic circulation and might contribute to its beneficial effects. J. Immunoassay Immunochem. 2021, 42, 252–264. [Google Scholar] [CrossRef]
- Lee, S.K.; Park, M.-J.; Jhun, J.Y.; Beak, J.-A.; Choi, J.W.; Rye, J.-Y.; Jang, J.W.; Bae, S.H.; Yoon, S.K.; Choi, H.J.; et al. Combination treatment with metformin and tacrolimus improves systemic immune cellular homeostasis by modulating Treg and Th17 imbalance. Front. Immunol. 2021, 11, 581728. [Google Scholar] [CrossRef]
- Borzouei, S.; Moghimi, H.; Zamani, A.; Behzad, M. Changes in T helper cell-related factors in patients with type 2 diabetes mellitus after empagliflozin therapy. Hum. Immunol. 2021, 82, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Wu, D.; Denroche, H.C.; Yao, Y.; Verchere, C.B.; Levings, M.K. IL-33 reverses an obesity-induced deficit in visceral adipose tissue ST2 + T regulatory cells and ameliorates adipose tissue inflammation and insulin resistance. J. Immunol. 2015, 194, 4777–4783. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Jing, J.; Peng, Z.; Liu, X.; Wang, X. Acacetin ameliorates insulin resistance in obesity mice through regulating Treg/Th17 balance via MiR-23b-3p/NEU1 axis. BMC Endocr. Disord. 2021, 21, 57. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.-K.; Yoon, B.-Y.; Jhun, J.-Y.; Oh, H.-J.; Kim, E.; Min, J.-K.; Cho, M.-L. Epigallocatechin-3-gallate ameliorates both obesity and autoinflammatory arthritis aggravated by obesity by altering the balance among CD4+ T-cell subsets. Immunol. Lett. 2014, 157, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Łuczyński, W.; Grubczak, K.; Moniuszko, M.; Głowińska-Olszewska, B.; Bossowski, A. Elevated levels of Th17 cells in children with central obesity. Scand. J. Clin. Lab. Investig. 2015, 75, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Schindler, T.I.; Wagner, J.-J.; Goedicke-Fritz, S.; Rogosch, T.; Coccejus, V.; Laudenbach, V.; Nikolaizik, W.; Härtel, C.; Maier, R.F.; Kerzel, S.; et al. TH17 cell frequency in peripheral blood is elevated in overweight children without chronic inflammatory diseases. Front. Immunol. 2017, 8, 1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.; Lichtenauer, M.; Strodthoff, D.; Winkels, H.; Wernly, B.; Bürger, C.; Kamchybekov, U.; Lutgens, E.; Figulla, H.-R.; Gerdes, N. Alterations in systemic levels of Th1, Th2, and Th17 cytokines in overweight adolescents and obese mice. Pediatr. Diabetes 2017, 18, 714–721. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Croce, S.; Avanzini, M.A.; Regalbuto, C.; Cordaro, E.; Vinci, F.; Zuccotti, G.; Calcaterra, V. Adipose Tissue Immunomodulation and Treg/Th17 Imbalance in the Impaired Glucose Metabolism of Children with Obesity. Children 2021, 8, 554. https://doi.org/10.3390/children8070554
Croce S, Avanzini MA, Regalbuto C, Cordaro E, Vinci F, Zuccotti G, Calcaterra V. Adipose Tissue Immunomodulation and Treg/Th17 Imbalance in the Impaired Glucose Metabolism of Children with Obesity. Children. 2021; 8(7):554. https://doi.org/10.3390/children8070554
Chicago/Turabian StyleCroce, Stefania, Maria Antonietta Avanzini, Corrado Regalbuto, Erika Cordaro, Federica Vinci, Gianvincenzo Zuccotti, and Valeria Calcaterra. 2021. "Adipose Tissue Immunomodulation and Treg/Th17 Imbalance in the Impaired Glucose Metabolism of Children with Obesity" Children 8, no. 7: 554. https://doi.org/10.3390/children8070554