
Fetal Fractures in an Infant with Maternal Ehlers-Danlos Syndrome, CCDC134 Pathogenic Mutation and a Negative Genetic Test for Osteogenesis Imperfecta


Abstract
:1. Introduction
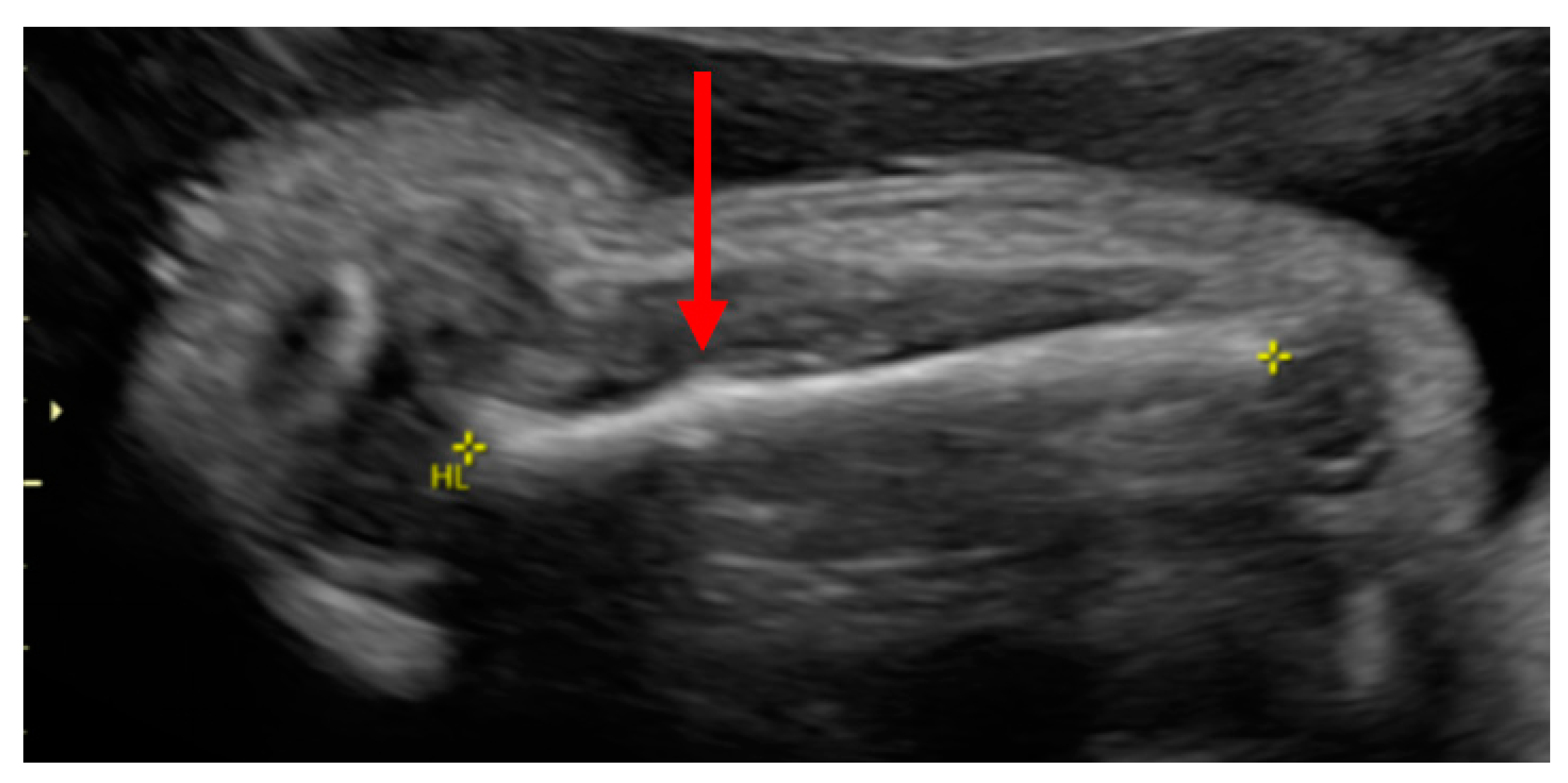
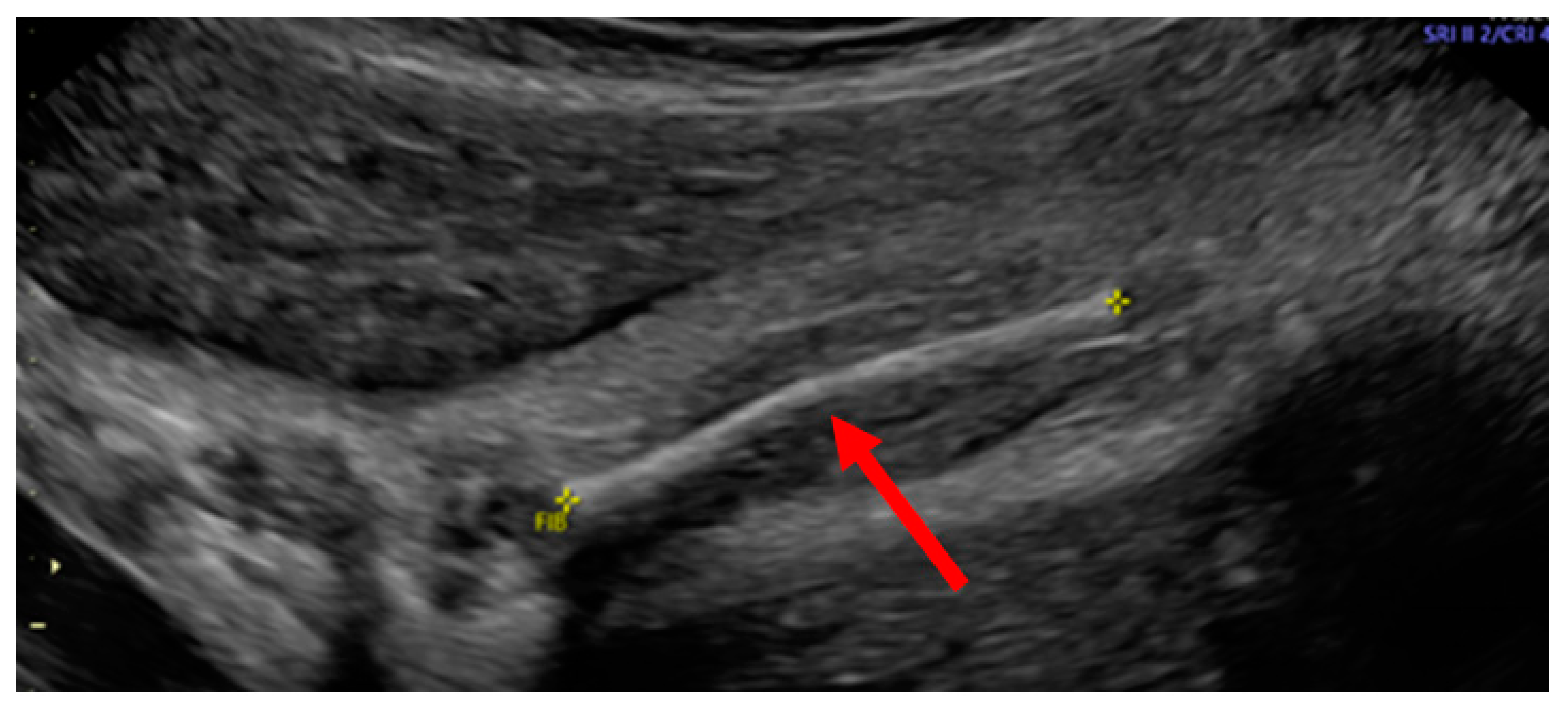
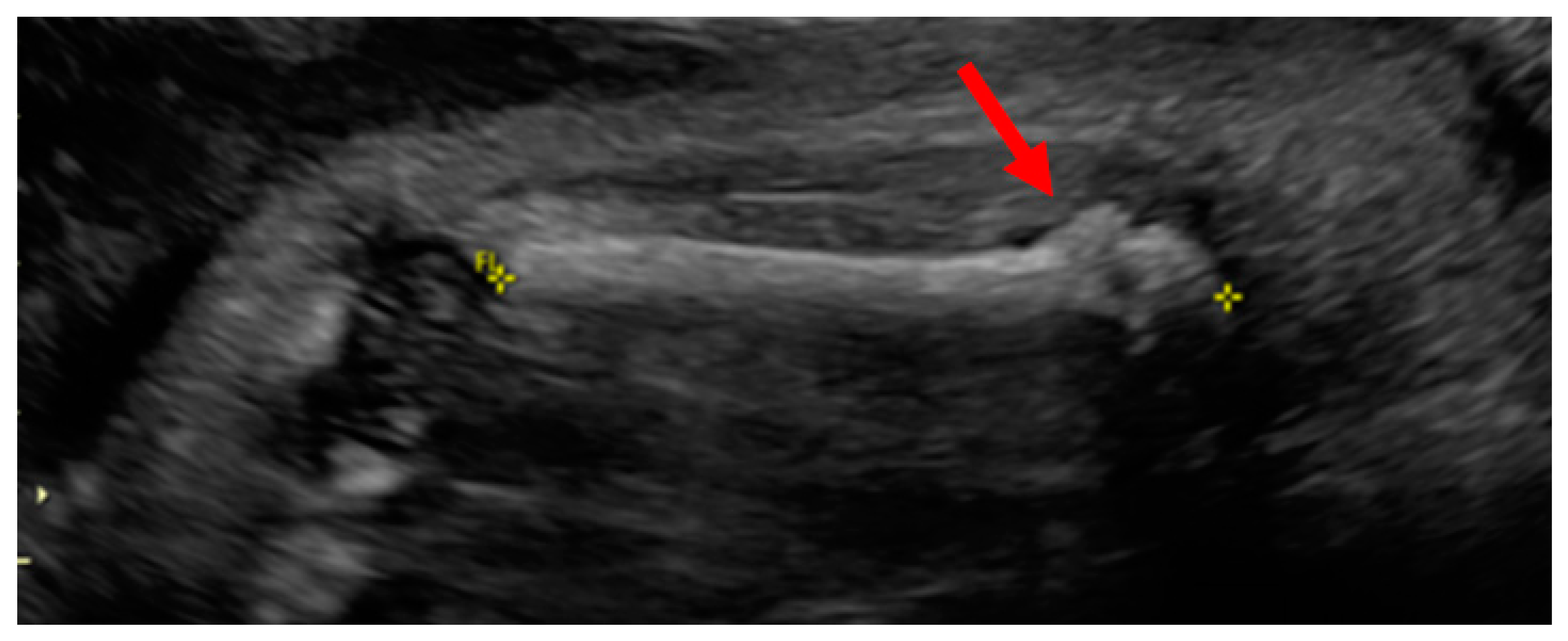
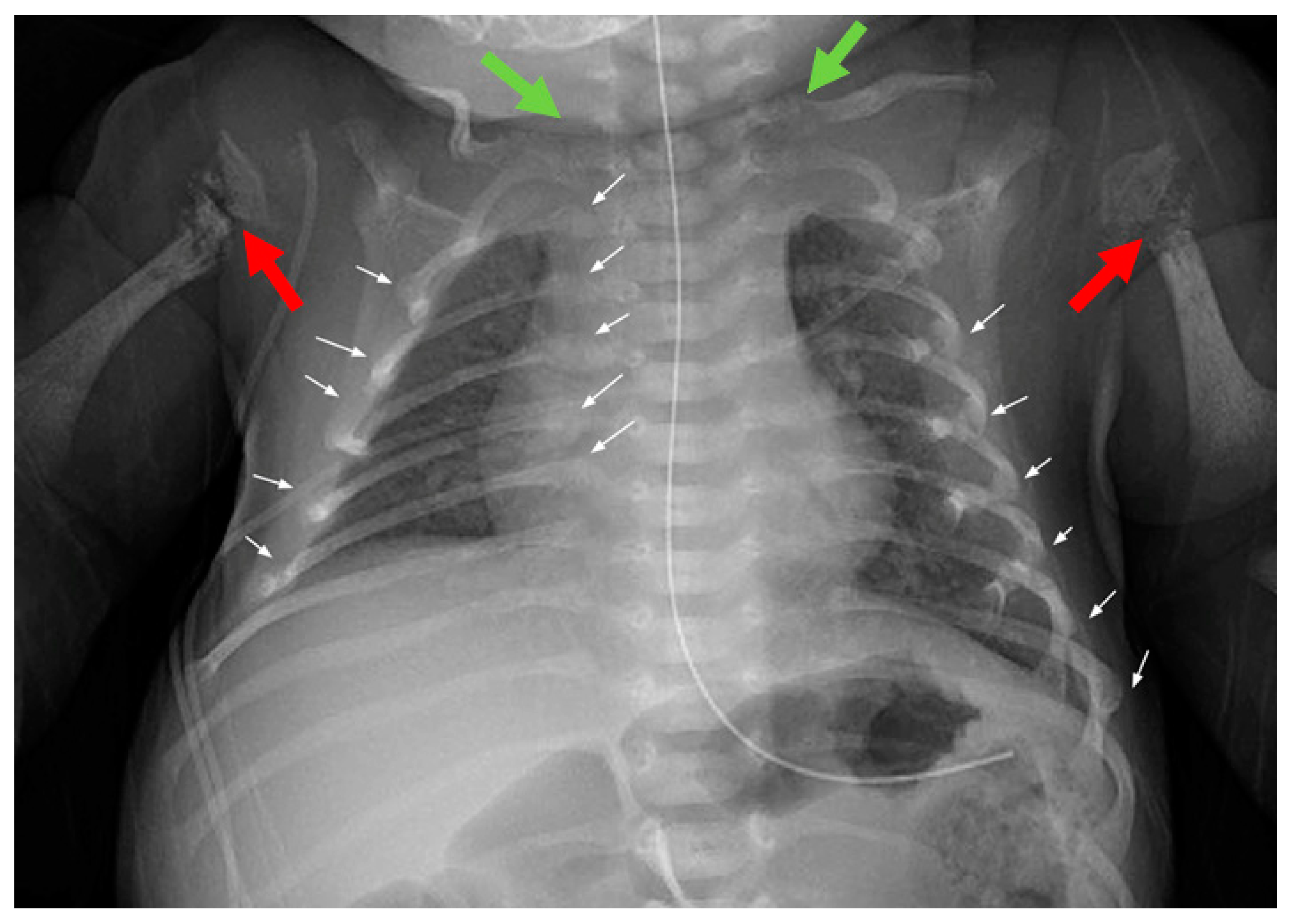
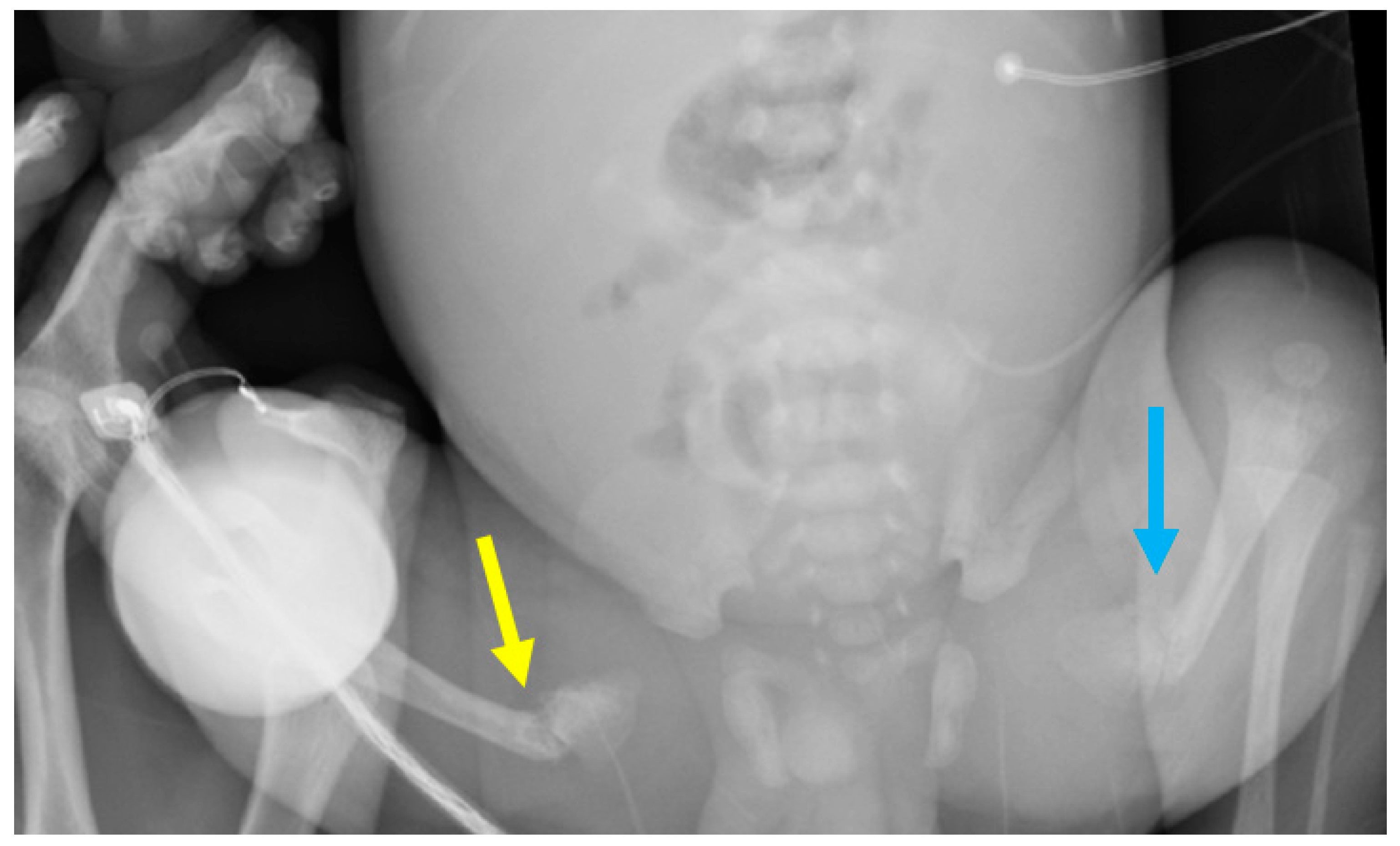
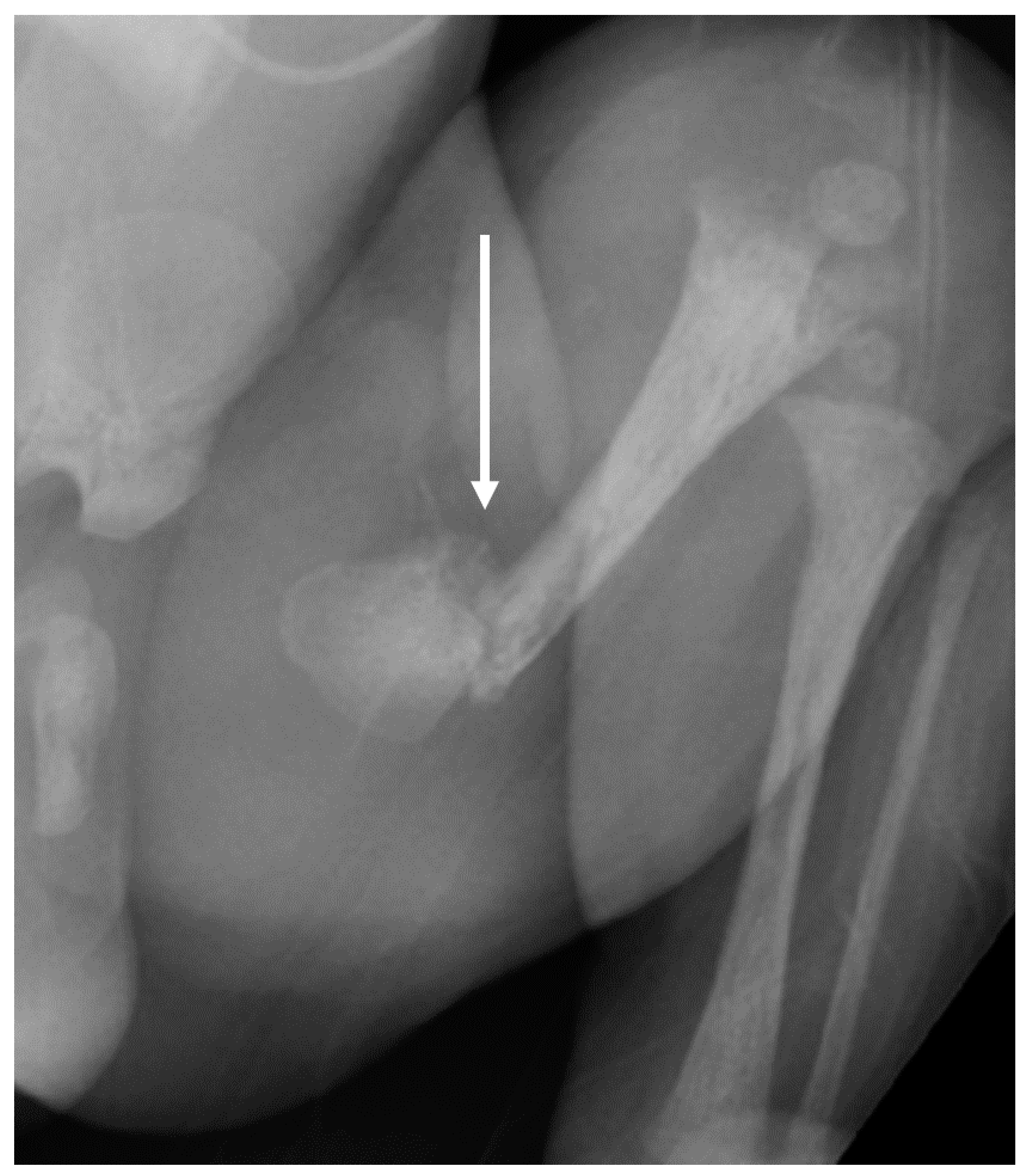
Case Report
2. Materials and Methods
Whole-Genome Sequencing
3. Results
3.1. Phenotypes of the Patient and Parents
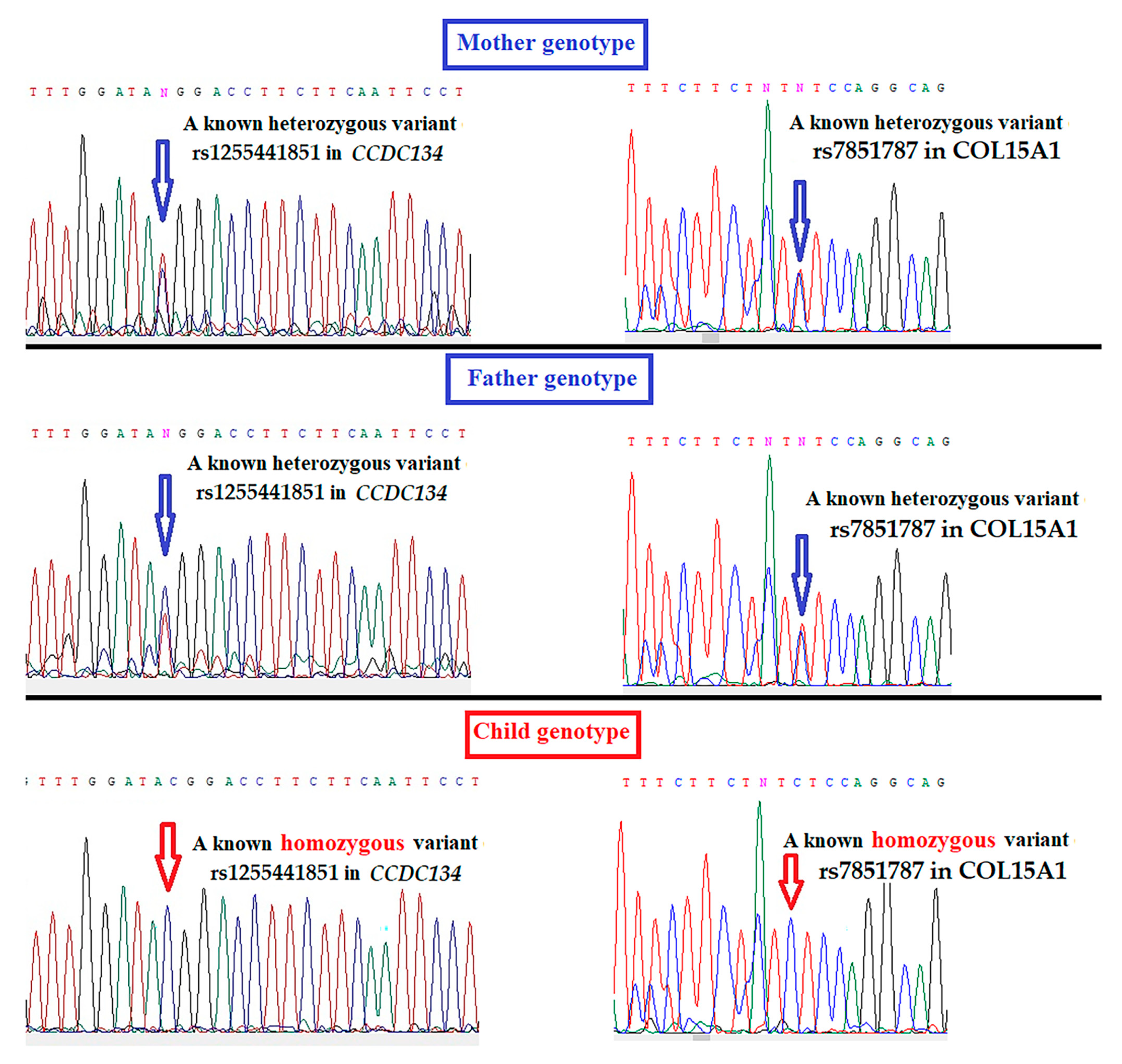
3.2. Whole-Genome Sequencing Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morgan, J.A.; Marcus, P.S. Prenatal Diagnosis and Management of Intrauterine Fracture. Obstet. Gynecol. Surv. 2010, 65, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Charoenngam, N.; Cevik, M.B.; Holick, M.F. Diagnosis and management of pediatric metabolic bone diseases associated with skeletal fragility. Curr. Opin. Pediatrics 2020, 32, 560–573. [Google Scholar] [CrossRef]
- Dubail, J.; Brunelle, P.; Baujat, G.; Huber, C.; Doyard, M.; Michot, C.; Chavassieux, P.; Khairouni, A.; Topouchian, V.; Monnot, S.; et al. Homozygous Loss-of-Function Mutations in CCDC134 Are Responsible for a Severe Form of Osteogenesis Imperfecta. J. Bone Miner. Res. 2020, 35, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Bardai, G.; Ward, L.M.; Trejo, P.; Moffatt, P.; Glorieux, F.H.; Rauch, F. Molecular diagnosis in children with fractures but no extraskeletal signs of osteogenesis imperfecta. Osteoporos. Int. 2017, 28, 2095–2101. [Google Scholar] [CrossRef]
- Gensemer, C.; Burks, R.; Kautz, S.; Judge, D.P.; Lavallee, M.; Norris, R.A. Hypermobile Ehlers-Danlos syndromes: Complex phenotypes, challenging diagnoses, and poorly understood causes. Dev. Dyn. 2021, 250, 318–344. [Google Scholar] [CrossRef]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Ritelli, M.; Colombi, M. Molecular insights in the pathogenesis of classical Ehlers-Danlos syndrome from transcriptome-wide expression profiling of patients’ skin fibroblasts. PLoS ONE 2019, 14, e0211647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beighton, P.; Paepe, A.D.; Steinmann, B.; Tsipouras, P.; Wenstrup, R.J. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Am. J. Med. Genet. 1998, 77, 31–37. [Google Scholar] [CrossRef]
- Kim, J.-M.; Yang, Y.-S.; Park, K.H.; Oh, H.; Greenblatt, M.B.; Shim, J.-H. The ERK MAPK Pathway Is Essential for Skeletal Development and Homeostasis. Int. J. Mol. Sci. 2019, 20, 1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Shi, T.; Ma, T.; Zhang, Y.; Ma, X.; Lu, Y.; Song, Q.; Liu, W.; Ma, D.; Qiu, X. CCDC134, a novel secretory protein, inhibits activation of ERK and JNK, but not p38 MAPK. Cell. Mol. Life Sci. 2007, 65, 338–349. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [Green Version]
- Lisignoli, G.; Lambertini, E.; Manferdini, C.; Gabusi, E.; Penolazzi, L.; Paolella, F.; Angelozzi, M.; Casagranda, V.; Piva, R. Collagen type XV and the ‘osteogenic status’. J. Cell. Mol. Med. 2017, 21, 2236–2244. [Google Scholar] [CrossRef] [Green Version]
- Tolkachov, A.; Fischer, C.; Ambrosi, T.H.; Bothe, M.; Han, C.-T.; Muenzner, M.; Mathia, S.; Salminen, M.; Seifert, G.; Thiele, M.; et al. Loss of the Hematopoietic Stem Cell Factor GATA2 in the Osteogenic Lineage Impairs Trabecularization and Mechanical Strength of Bone. Mol. Cell. Biol. 2018, 38, e00599-17. [Google Scholar] [CrossRef] [Green Version]
- Schiaffino, S.; Rossi, A.C.; Smerdu, V.; Leinwand, L.A.; Reggiani, C. Developmental myosins: Expression patterns and functional significance. Skelet. Muscle 2015, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Toydemir, R.M.; Rutherford, A.; Whitby, F.G.; Jorde, L.B.; Carey, J.C.; Bamshad, M.J. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat. Genet. 2006, 38, 561–565. [Google Scholar] [CrossRef]
- Zieba, J.; Zhang, W.; Chong, J.X.; Forlenza, K.N.; Martin, J.H.; Heard, K.; Grange, D.K.; Butler, M.G.; Kleefstra, T.; Lachman, R.S.; et al. A postnatal role for embryonic myosin revealed by MYH3 mutations that alter TGFβ signaling and cause autosomal dominant spondylocarpotarsal synostosis. Sci. Rep. 2017, 7, srep41803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, M.; Kauschke, V.; Sender, J.; Kampschulte, M.; Kovtun, A.; Dürselen, L.; Heiss, C.; Lips, K.S. Bone status of adult female butyrylcholinesterase gene-deficient mice. Int. Immunopharmacol. 2015, 29, 208–214. [Google Scholar] [CrossRef]
- Palumbo, P.; Di Muro, E.; Accadia, M.; Benvenuto, M.; Di Giacomo, M.; Castellana, S.; Mazza, T.; Castori, M.; Palumbo, O.; Carella, M. Whole Exome Sequencing Reveals a Novel AUTS2 In-Frame Deletion in a Boy with Global Developmental Delay, Absent Speech, Dysmorphic Features, and Cerebral Anomalies. Genes 2021, 12, 229. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [Green Version]
- Colombi, M.; Dordoni, C.; Chiarelli, N.; Ritelli, M. Differential diagnosis and diagnostic flow chart of joint hypermobility syndrome/ehlers-danlos syndrome hypermobility type compared to other heritable connective tissue disorders. Am. J. Med. Genet. Part C Semin. Med. Genet. 2015, 169, 6–22. [Google Scholar] [CrossRef] [PubMed]
- Mazziotti, G.; Dordoni, C.; Doga, M.; Galderisi, F.; Venturini, M.; Calzavara-Pinton, P.; Maroldi, R.; Giustina, A.; Colombi, M. High prevalence of radiological vertebral fractures in adult patients with Ehlers–Danlos syndrome. Bone 2016, 84, 88–92. [Google Scholar] [CrossRef] [Green Version]
- Holick, M.F.; Hossein-Nezhad, A.; Tabatabaei, F. Multiple fractures in infants who have Ehlers-Danlos/hypermobility syndrome and or vitamin D deficiency: A case series of 72 infants whose parents were accused of child abuse and neglect. Derm. -Endocrinol. 2017, 9, e1279768. [Google Scholar] [CrossRef]
- Rolfes, M.C.; Deyle, D.R.; King, K.S.; Hand, J.L.; Graff, A.H.; Derauf, C. Fracture incidence in Ehlers-Danlos syndrome—A population-based case-control study. Child Abus. Negl. 2019, 91, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Shur, N.; Carey, J.C. Genetic differentials of child abuse: Is your case rare or real? Am. J. Med. Genet. Part C Semin. Med. Genet. 2015, 169, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Tofts, L.J.; Elliott, E.J.; Munns, C.; Pacey, V.; Sillence, D. The differential diagnosis of children with joint hypermobility: A review of the literature. Pediatr. Rheumatol. 2009, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamifar, H.; Ilkhanipoor, H.; Ajami, G.; Karamizadeh, Z.; Amirhakimi, G.; Shakiba, A.-M. Cardiovascular Involvement in Children with Osteogenesis Imperfecta. Iran. J. Pediatr. 2013, 23, 513–518. [Google Scholar] [PubMed]
- Morlino, S.; Micale, L.; Ritelli, M.; Rohrbach, M.; Zoppi, N.; Vandersteen, A.; Mackay, S.; Agolini, E.; Cocciadiferro, D.; Sasaki, E.; et al. COL1-related overlap disorder: A novel connective tissue disorder incorporating the osteogenesis imperfecta/Ehlers-Danlos syndrome overlap. Clin. Genet. 2019, 97, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Shur, N.; Robin, N.H. Editorial: Medical genetics, expert medical testimony, and suspected child abuse cases: A call for evidence-based standards in clinic and the courtroom. Curr. Opin. Pediatr. 2021, 33, 1–2. [Google Scholar] [CrossRef]
- George, M.P.; Shur, N.E.; Peréz-Rosselló, J.M. Ehlers–Danlos syndrome: What the radiologist needs to know. Pediatr. Radiol. 2021, 51, 1023–1028. [Google Scholar] [CrossRef]
- Shur, N.E.; Summerlin, M.L.; McIntosh, B.J.; Shalaby-Rana, E.; Hinds, T.S. Genetic causes of fractures and subdural hematomas: Fact versus fiction. Pediatr. Radiol. 2021, 51, 1029–1043. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mother | Father | Infant | |
|---|---|---|---|
| Phenotype | hEDS and nonunion fracture | Normal | Intrauterine Fractures |
| Gene | Genotypes | Genotypes | Genotypes |
| CCDC134 | N/V | N/V | V/V |
| COL15A1 | N/V | N/V | V/V |
| ZFPM1 | N/V | N/V | V/V |
| SNTB1 | N/V | N/V | V/V |
| F13B | N/N | N/V | N/V |
| TTN | N/N | N/V | N/V |
| ACAD9 | N/V | N/N | N/V |
| BCHE | N/N | N/V | N/V |
| NBEAL2 | N/N | N/V | N/V |
| AUTS2 | N/V | N/N | N/V |
| ASPH | N/V | N/N | N/V |
| NCAPD3 | N/N | N/V | N/V |
| CABP4 | N/N | N/V | N/V |
| MYH3 | N/V | N/N | N/V |
| KIR2DL3 | N/N | N/V | N/V |
| CRYBB3 | N/V | N/N | N/V |
| ZNF75D | N/V | N/N | N/V |
| Zygosity of Variant in the Infant | Gene | SNP | HGVSC | HGVSP | Allele Frequency | Allele | Clinical Significance | Consequence | Suggested Gene Function | SIFT | PolyPhen |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Homozygous variants | CCDC134 *a | rs1255441851 | c.2T>C | p.Met1Thr | 0.000004 | T > C | Not reported | Initiator codon loss-of-function | Regulating ERK-MAPK pathwayOsteogenesis Imperfecta phenotype | Deleterious | Probably damaging |
| COL15A1 * | rs7851787 | c.1762-6T>C | - | 0.15361 | T > C | Not reported | Splice region variant | Bone extracellular matrix protein | - | - | |
| ZFPM1 * | rs759189176 | c.1335_1338del | p.Leu446fs | 0.0003 | delTCTG | Not reported | Frameshift variant | Regulating osteogenic lineage by interaction with GATA2 | |||
| SNTB1 | rs547154887 | c.12_14GGC | p.Ala8_Ala10del | 0.02 | delGCC | Not reported | Inframe deletion | Unknown | - | - | |
| Heterozygous variants | F13B | rs17514281 | c.1025T>C | p.Ile342Thr | 0.007 | A > G | Conflicting | Missense variant | Unknown | Deleterious | Probably damaging |
| TTN | rs397517630 | c.57586C>G | p.Leu19196Val | 0.00017 | G > C | Conflicting | Missense variant | Unknown | - | Probably damaging | |
| ACAD9 | rs863224844 | c.359del | p.Phe120fs | 0.0001 | delT | Likely pathogenic | Frameshift variant | Unknown | - | - | |
| BCHE * | rs1799807 | c.293A>G | p.Asp98Gly | 0.012 | T > C | Likely pathogenic | Missense variant | Regulating the number of osteoclasts and bone microarchitecture | Deleterious | Probably damaging | |
| NBEAL2 | rs201373710 | c.1948G>A | p.Gly650Arg | 0.0016 | G > A | Conflicting | Missense variant | Bone marrow | Deleterious | Probably damaging | |
| AUTS2 * | rs767529359 | c.1295C>T | p.Pro432Leu | 0.0002 | C > A | Conflicting | Missense variant | Skeletal anomalies | Deleterious | Probably damaging | |
| ASPH | rs80163539 | c.518del | p.Asp173fs | 0.001 | delT | Conflicting | Frameshift variant | Osteogenic differentiationunknown | - | - | |
| NCAPD3 | rs151013524 | c.1981G>T | p.Asp661Tyr | 0.0019 | C > A | Uncertain significance | Missense variant | Abnormal development of lower spine | Deleterious | Probably damaging | |
| CABP4 | rs146764702 | c.547G>C | p.Gly183Arg | 0.0005 | G > C | Uncertain significance | Missense variant | Unknown | Deleterious | Probably damaging | |
| MYH3 * | rs557849165 | c.-9+1G>A | - | 0.00274 | C > T | Pathogenic | Splice donor variant | Skeletal dysplasia | - | - | |
| KIR2DL3 | rs193921051 | c.71-4C>T | - | 0.08 | C > T | Uncertain significance | Splice region variant | Bone marrow | - | - | |
| CRYBB3 | rs147937174 | c.584G>A | p.Arg195His | 0.0001 | G > A | Conflicting * | Missense variant | Unknown | Deleterious | Probably damaging | |
| Hemizygous variants | ZNF75D | rs150700463 | c.934C>T | p.Gln312Ter | 0.00134 | G > A | Not reported | Stop gained | Unknown | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holick, M.F.; Shirvani, A.; Charoenngam, N. Fetal Fractures in an Infant with Maternal Ehlers-Danlos Syndrome, CCDC134 Pathogenic Mutation and a Negative Genetic Test for Osteogenesis Imperfecta. Children 2021, 8, 512. https://doi.org/10.3390/children8060512
Holick MF, Shirvani A, Charoenngam N. Fetal Fractures in an Infant with Maternal Ehlers-Danlos Syndrome, CCDC134 Pathogenic Mutation and a Negative Genetic Test for Osteogenesis Imperfecta. Children. 2021; 8(6):512. https://doi.org/10.3390/children8060512
Chicago/Turabian StyleHolick, Michael F., Arash Shirvani, and Nipith Charoenngam. 2021. "Fetal Fractures in an Infant with Maternal Ehlers-Danlos Syndrome, CCDC134 Pathogenic Mutation and a Negative Genetic Test for Osteogenesis Imperfecta" Children 8, no. 6: 512. https://doi.org/10.3390/children8060512