
A Novel Variant in the TP53 Gene Causing Li–Fraumeni Syndrome
 ,
,  , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Case Report
2.1. Presentation
2.2. Hormonal Profile
2.3. Surgical Treatment
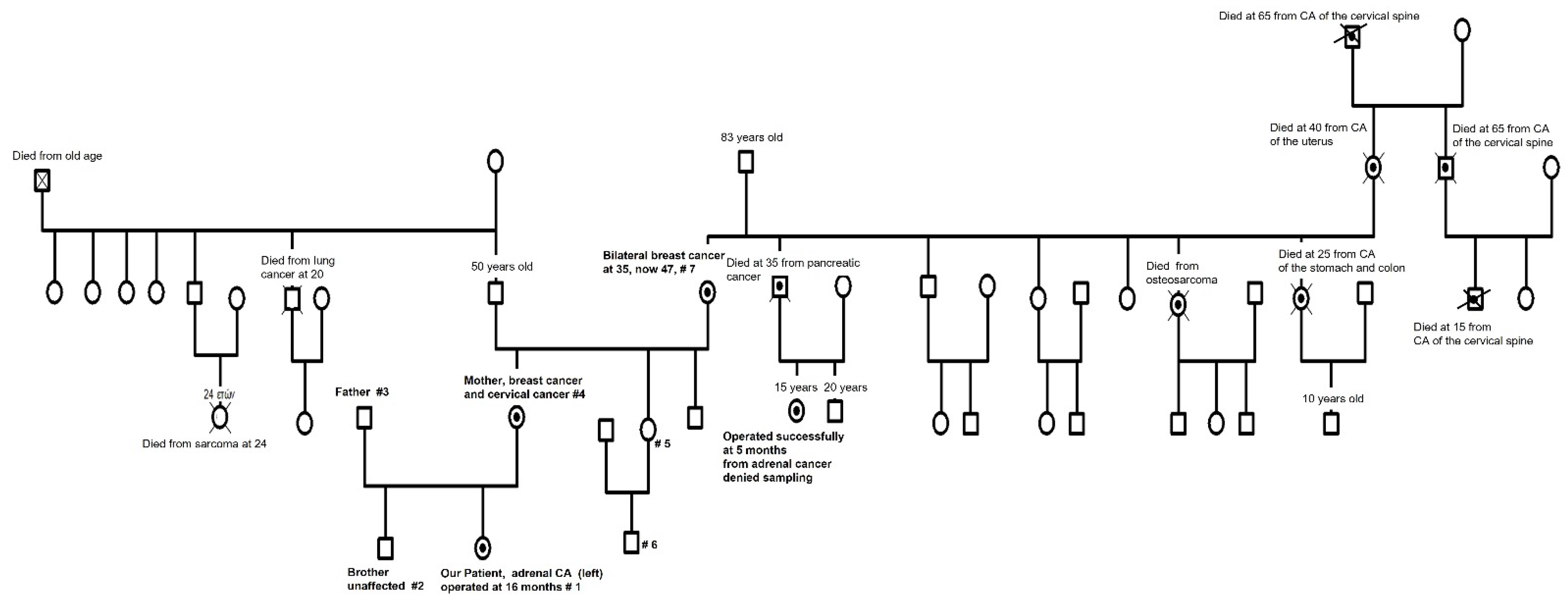
2.4. Family History—Ordering Whole-Exome Sequencing
2.5. Whole-Exome Sequencing (WES)
2.6. Detection of Breast Cancer in the Patient’s Mother
2.7. Detection of Cervical Cancer in the Patient’s Mother
2.8. Detection of Osteosarcoma in Our Patient
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levine, A.J. Spontaneous and inherited TP53 genetic alterations. Oncogene 2021, 40, 5975–5983. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.J.; Dodd-Eaton, E.B.; Peng, G.; Bojadzieva, J.; Chen, J.; Amos, C.I.; Frone, M.N.; Khincha, P.P.; Mai, P.L.; Savage, S.A.; et al. Penetrance of Different Cancer Types in Families with Li-Fraumeni Syndrome: A Validation Study Using Multicenter Cohorts. Cancer Res. 2020, 80, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Aedma, S.K.; Kasi, A. Li-Fraumeni Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Consul, N.; Amini, B.; Ibarra-Rovira, J.J.; Blair, K.J.; Moseley, T.W.; Taher, A.; Shah, K.B.; Elsayes, K.M. Li-Fraumeni Syndrome and Whole-Body MRI Screening: Screening Guidelines, Imaging Features, and Impact on Patient Management. AJR Am. J. Roentgenol. 2021, 216, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. The Evolution of Tumor Formation in Humans and Mice with Inherited Mutations in the p53 Gene. Curr. Top. Microbiol. Immunol. 2017, 407, 205–221. [Google Scholar] [CrossRef]
- Kleinerman, R.A. Radiation-sensitive genetically susceptible pediatric sub-populations. Pediatr. Radiol. 2009, 39 (Suppl. S1), S27–S31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisada, M.; Garber, J.E.; Fung, C.Y.; Fraumeni, J.F., Jr.; Li, F.P. Multiple primary cancers in families with Li-Fraumeni syndrome. J. Natl. Cancer Inst. 1998, 90, 606–611. [Google Scholar] [CrossRef]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Xiang, J.; Peng, J.; Baxter, S.; Peng, Z. AutoPVS1: An automatic classification tool for PVS1 interpretation of null variants. Human Mutat. 2020, 41, 1488–1498. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Human Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Olivier, M.; Goldgar, D.E.; Sodha, N.; Ohgaki, H.; Kleihues, P.; Hainaut, P.; Eeles, R.A. Li-Fraumeni and related syndromes: Correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003, 63, 6643–6650. [Google Scholar]
- Gonzalez, K.D.; Noltner, K.A.; Buzin, C.H.; Gu, D.; Wen-Fong, C.Y.; Nguyen, V.Q.; Han, J.H.; Lowstuter, K.; Longmate, J.; Sommer, S.S.; et al. Beyond Li Fraumeni Syndrome: Clinical characteristics of families with p53 germline mutations. J. Clin. Oncol. 2009, 27, 1250–1256. [Google Scholar] [CrossRef]
- Ruijs, M.W.; Verhoef, S.; Rookus, M.A.; Pruntel, R.; van der Hout, A.H.; Hogervorst, F.B.; Kluijt, I.; Sijmons, R.H.; Aalfs, C.M.; Wagner, A.; et al. TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: Mutation detection rate and relative frequency of cancers in different familial phenotypes. J. Med. Genet. 2010, 47, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.M.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugières, L.; et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J. Clin. Oncol. 2015, 33, 2345–2352. [Google Scholar] [CrossRef]
- Chompret, A.; Abel, A.; Stoppa-Lyonnet, D.; Brugiéres, L.; Pagés, S.; Feunteun, J.; Bonaïti-Pellié, C. Sensitivity and predictive value of criteria for p53 germline mutation screening. J. Med. Genet. 2001, 38, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Chompret, A.; Brugières, L.; Ronsin, M.; Gardes, M.; Dessarps-Freichey, F.; Abel, A.; Hua, D.; Ligot, L.; Dondon, M.G.; Bressac-de Paillerets, B.; et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br. J. Cancer 2000, 82, 1932–1937. [Google Scholar] [CrossRef] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017, 2017, PO.17.00029. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Malkin, D. Li-fraumeni syndrome. Genes Cancer 2011, 2, 475–484. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Gillam, L.; Visvanathan, K.; Hansford, J.R.; McCarthy, M.C. Clinical Utility of Precision Medicine in Pediatric Oncology: A Systematic Review. JCO Precis. Oncol. 2021, 5, 1088–1102. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.S.; de Haes, J.C. Denial in cancer patients, an explorative review. Psycho-Oncology 2007, 16, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Storm, C.; Agarwal, R.; Offit, K. Ethical and legal implications of cancer genetic testing: Do physicians have a duty to warn patients’ relatives about possible genetic risks? J. Oncol. Pract. 2008, 4, 229–230. [Google Scholar] [CrossRef] [Green Version]
- Stratakis, C.A. Genetics and the New (Precision) Medicine and Endocrinology: In Medias Res or Ab Initio? Endocrinol. Metab. Clin. N. Am. 2017, 46, xv–xvi. [Google Scholar] [CrossRef]
- Tran, A.; Klossner, Q.; Crain, T.; Prasad, V. Shifting, overlapping and expanding use of “precision oncology” terminology: A retrospective literature analysis. BMJ Open 2020, 10, e036357. [Google Scholar] [CrossRef]
- Capasso, M.; Montella, A.; Tirelli, M.; Maiorino, T.; Cantalupo, S.; Iolascon, A. Genetic Predisposition to Solid Pediatric Cancers. Front. Oncol. 2020, 10, 590033. [Google Scholar] [CrossRef]
- Wasserman, J.D.; Novokmet, A.; Eichler-Jonsson, C.; Ribeiro, R.C.; Rodriguez-Galindo, C.; Zambetti, G.P.; Malkin, D. Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: A children’s oncology group study. J. Clin. Oncol. 2015, 33, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Sweet-Cordero, E.A.; Biegel, J.A. The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science 2019, 363, 1170–1175. [Google Scholar] [CrossRef]
- Savary, C.; Kim, A.; Lespagnol, A.; Gandemer, V.; Pellier, I.; Andrieu, C.; Pagès, G.; Galibert, M.-D.; Blum, Y.; de Tayrac, M. Depicting the genetic architecture of pediatric cancers through an integrative gene network approach. Sci. Rep. 2020, 10, 1224. [Google Scholar] [CrossRef] [Green Version]
- Diets, I.J.; Waanders, E.; Ligtenberg, M.J.; van Bladel, D.A.G.; Kamping, E.J.; Hoogerbrugge, P.M.; Hopman, S.; Olderode-Berends, M.J.; Gerkes, E.H.; Koolen, D.A.; et al. High Yield of Pathogenic Germline Mutations Causative or Likely Causative of the Cancer Phenotype in Selected Children with Cancer. Clin. Cancer Res. 2018, 24, 1594–1603. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Gene | Disease | Mode of Inheritance | Variant | Coding DNA | Zygosity | Inherited from | Classification |
|---|---|---|---|---|---|---|---|
| TP53 | Li–Fraumeni Syndrome | Autosomal Dominant | p.E298FfsX48 | c.892delGinsTIT | Heterozygous | Proband’s mother and maternal grandmother | Likely Pathogenic Variant |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadimitriou, D.T.; Stratakis, C.A.; Kattamis, A.; Glentis, S.; Dimitrakakis, C.; Spyridis, G.P.; Christopoulos, P.; Mastorakos, G.; Vlahos, N.F.; Iacovidou, N. A Novel Variant in the TP53 Gene Causing Li–Fraumeni Syndrome. Children 2023, 10, 1150. https://doi.org/10.3390/children10071150
Papadimitriou DT, Stratakis CA, Kattamis A, Glentis S, Dimitrakakis C, Spyridis GP, Christopoulos P, Mastorakos G, Vlahos NF, Iacovidou N. A Novel Variant in the TP53 Gene Causing Li–Fraumeni Syndrome. Children. 2023; 10(7):1150. https://doi.org/10.3390/children10071150
Chicago/Turabian StylePapadimitriou, Dimitrios T., Constantine A. Stratakis, Antonis Kattamis, Stavros Glentis, Constantine Dimitrakakis, George P. Spyridis, Panagiotis Christopoulos, George Mastorakos, Nikolaos F. Vlahos, and Nicoletta Iacovidou. 2023. "A Novel Variant in the TP53 Gene Causing Li–Fraumeni Syndrome" Children 10, no. 7: 1150. https://doi.org/10.3390/children10071150