
Role of UPR Sensor Activation in Cell Death–Survival Decision of Colon Cancer Cells Stressed by DPE Treatment

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Treatments
2.2. Cell Viability
2.3. Cell Proliferation
2.4. Cell Cycle Analysis
2.5. Western Blot Analysis
2.6. Antibodies
2.7. Statistical Analysis
3. Results
3.1. DPE Impairs Cell Survival and Proliferation in a Dose-Dependent Fashion in WT and Mutp53-Carrying Colon Cancer Cells While Sparing Normal Colon Epithelial Cells
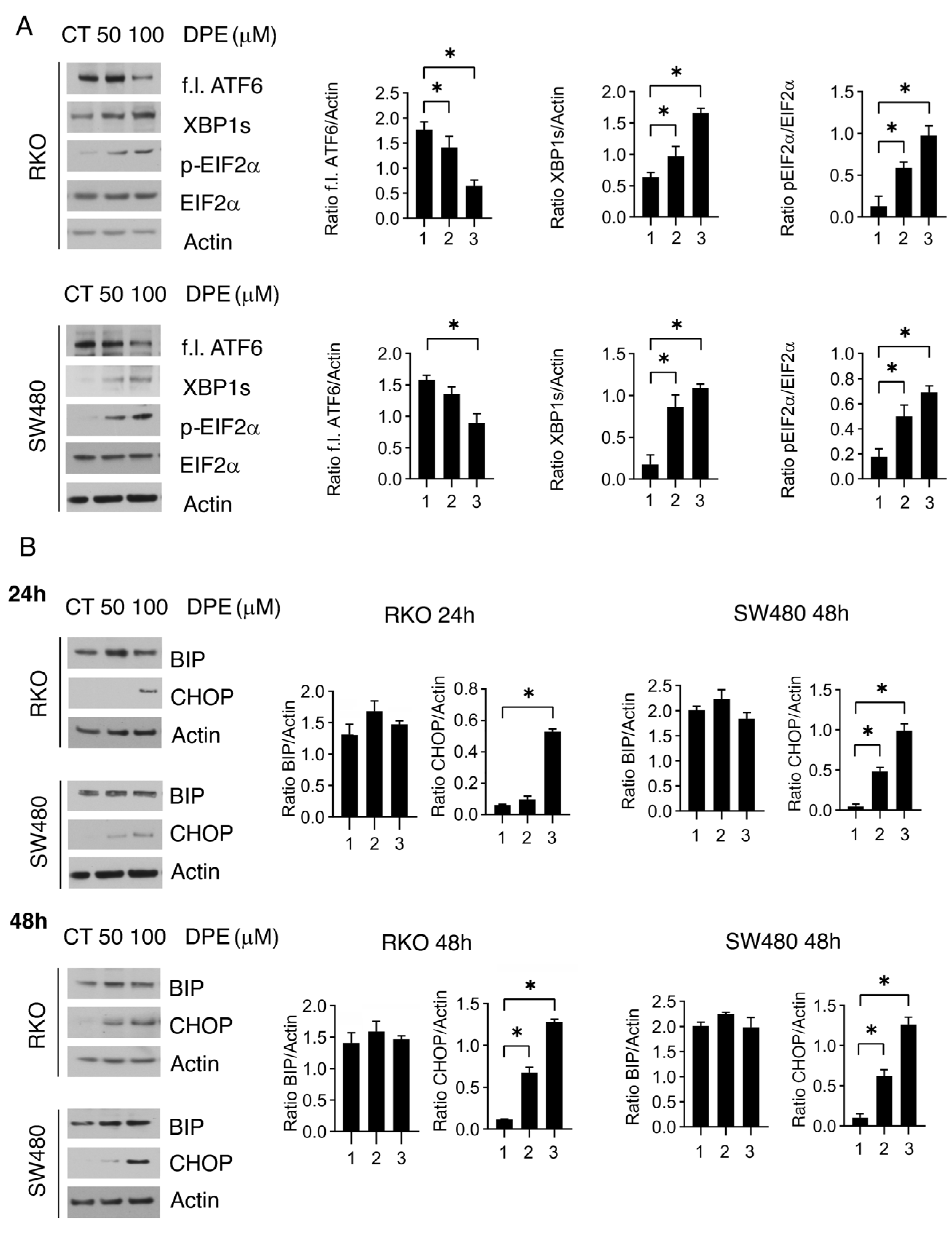
3.2. DPE Activated All UPR Branches and Induced Dose-Dependent Upregulation of CHOP
3.3. DPE Induces ERK1/2 De-Phosphorylation and DUSP5 Upregulation
3.4. BiP Is Upregulated in the Fraction of Colon Cancer Cells Resistant to DPE and Targeting BiP May Improve the Outcome of DPE-Treatment
3.5. UPR Sensor Inhibition Differently Affects Cell Survival in DPE-Treated Colon Cancer Cells
3.6. GSK2606414 Downregulates CHOP and Upregulates Mcl-1 While 4u8c Induces Opposite Effects
3.7. Atf6 Inhibition by Ceapin Reduces Bip Expression Levels Both in Rko and sw480 Cells and Stabilizes wtp53 in the Former While Downregulating mutp53 in the Latter
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Dong, D.; Ni, M.; Li, J.; Xiong, S.; Ye, W.; Virrey, J.J.; Mao, C.; Ye, R.; Wang, M.; Pen, L.; et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008, 68, 498–505. [Google Scholar] [CrossRef] [Green Version]
- Cerezo, M.; Benhida, R.; Rocchi, S. Targeting BIP to induce Endoplasmic Reticulum stress and cancer cell death. Oncoscience 2016, 3, 306–307. [Google Scholar] [CrossRef] [PubMed]
- Virrey, J.J.; Dong, D.; Stiles, C.; Patterson, J.B.; Pen, L.; Ni, M.; Schonthal, A.H.; Chen, T.C.; Hofman, F.M.; Lee, A.S. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol. Cancer Res. 2008, 6, 1268–1275. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.E. Killing cancer cells, twice with one shot. Cell Death Differ. 2014, 21, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Ayyoub, M.; Routy, B.; Kroemer, G. Microbiome and Anticancer Immunosurveillance. Cell 2016, 165, 276–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilardini Montani, M.S.; Benedetti, R.; Piconese, S.; Pulcinelli, F.M.; Timperio, A.M.; Romeo, M.A.; Masuelli, L.; Mattei, M.; Bei, R.; D’Orazi, G.; et al. PGE2 Released by Pancreatic Cancer Cells Undergoing ER Stress Transfers the Stress to DCs Impairing Their Immune Function. Mol. Cancer Ther. 2021, 20, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Martucciello, S.; Masullo, M.; Cerulli, A.; Piacente, S. Natural Products Targeting ER Stress, and the Functional Link to Mitochondria. Int. J. Mol. Sci. 2020, 21, 1905. [Google Scholar] [CrossRef] [Green Version]
- Cirone, M.; Di Renzo, L.; Lotti, L.V.; Conte, V.; Trivedi, P.; Santarelli, R.; Gonnella, R.; Frati, L.; Faggioni, A. Activation of dendritic cells by tumor cell death. Oncoimmunology 2012, 1, 1218–1219. [Google Scholar] [CrossRef] [Green Version]
- Guichard, C.; Pedruzzi, E.; Fay, M.; Marie, J.C.; Braut-Boucher, F.; Daniel, F.; Grodet, A.; Gougerot-Pocidalo, M.A.; Chastre, E.; Kotelevets, L.; et al. Dihydroxyphenylethanol induces apoptosis by activating serine/threonine protein phosphatase PP2A and promotes the endoplasmic reticulum stress response in human colon carcinoma cells. Carcinogenesis 2006, 27, 1812–1827. [Google Scholar] [CrossRef] [Green Version]
- Martinez, L.; Ros, G.; Nieto, G. Hydroxytyrosol: Health Benefits and Use as Functional Ingredient in Meat. Medicines 2018, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Strate, L.L.; Syngal, S. Hereditary colorectal cancer syndromes. Cancer Causes Control 2005, 16, 201–213. [Google Scholar] [CrossRef]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, J.; Arenzana, N.; Tirasophon, W.; Kaufman, R.J.; Prywes, R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J. Biol. Chem. 2000, 275, 27013–27020. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Jo, H.J.; Yang, J.W.; Park, J.H.; Choi, E.S.; Lim, C.S.; Lee, S.; Han, C.Y. Endoplasmic Reticulum Stress Increases DUSP5 Expression via PERK-CHOP Pathway, Leading to Hepatocyte Death. Int. J. Mol. Sci. 2019, 20, 4369. [Google Scholar] [CrossRef] [Green Version]
- Buffet, C.; Hecale-Perlemoine, K.; Bricaire, L.; Dumont, F.; Baudry, C.; Tissier, F.; Bertherat, J.; Cochand-Priollet, B.; Raffin-Sanson, M.L.; Cormier, F.; et al. DUSP5 and DUSP6, two ERK specific phosphatases, are markers of a higher MAPK signaling activation in BRAF mutated thyroid cancers. PLoS ONE 2017, 12, e0184861. [Google Scholar] [CrossRef] [PubMed]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Bougie, P.; Halliez, M.; Moreau, P.; Pellat-Deceunynck, C.; Amiot, M. Repression of Mcl-1 and disruption of the Mcl-1/Bak interaction in myeloma cells couple ER stress to mitochondrial apoptosis. Cancer Lett. 2016, 383, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- Meares, G.P.; Liu, Y.; Rajbhandari, R.; Qin, H.; Nozell, S.E.; Mobley, J.A.; Corbett, J.A.; Benveniste, E.N. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol. Cell Biol. 2014, 34, 3911–3925. [Google Scholar] [CrossRef] [Green Version]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: A possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Yang, H.; Niemeijer, M.; van de Water, B.; Beltman, J.B. ATF6 Is a Critical Determinant of CHOP Dynamics during the Unfolded Protein Response. iScience 2020, 23, 100860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Sicari, D.; Fantuz, M.; Bellazzo, A.; Valentino, E.; Apollonio, M.; Pontisso, I.; Di Cristino, F.; Dal Ferro, M.; Bicciato, S.; Del Sal, G.; et al. Mutant p53 improves cancer cells’ resistance to endoplasmic reticulum stress by sustaining activation of the UPR regulator ATF6. Oncogene 2019, 38, 6184–6195. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Beaver, J.M.; Liu, Y.; Zhang, Z. Dynamics of p53: A Master Decider of Cell Fate. Genes 2017, 8, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Ojha, R.; Amaravadi, R.K. Targeting the unfolded protein response in cancer. Pharmacol. Res. 2017, 120, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A. Regulation of Cytokine Production by the Unfolded Protein Response; Implications for Infection and Autoimmunity. Front. Immunol. 2018, 9, 422. [Google Scholar] [CrossRef] [PubMed]
- Gonnella, R.; Gilardini Montani, M.S.; Guttieri, L.; Romeo, M.A.; Santarelli, R.; Cirone, M. IRE1 Alpha/XBP1 Axis Sustains Primary Effusion Lymphoma Cell Survival by Promoting Cytokine Release and STAT3 Activation. Biomedicines 2021, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, P.G.; Mazon, M.J.; Eraso, P.; Portillo, F. UPR: An Upstream Signal to EMT Induction in Cancer. J. Clin. Med. 2019, 8, 624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santarelli, R.; Arteni, A.M.B.; Gilardini Montani, M.S.; Romeo, M.A.; Gaeta, A.; Gonnella, R.; Faggioni, A.; Cirone, M. KSHV dysregulates bulk macroautophagy, mitophagy and UPR to promote endothelial to mesenchymal transition and CCL2 release, key events in viral-driven sarcomagenesis. Int. J. Cancer 2020, 147, 3500–3510. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Investig. 2012, 122, 4621–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [Green Version]
- D’Orazi, G.; Cirone, M. Mutant p53 and Cellular Stress Pathways: A Criminal Alliance That Promotes Cancer Progression. Cancers 2019, 11, 614. [Google Scholar] [CrossRef] [Green Version]
- D’Orazi, G.; Cordani, M.; Cirone, M. Oncogenic pathways activated by pro-inflammatory cytokines promote mutant p53 stability: Clue for novel anticancer therapies. Cell Mol. Life Sci. 2021, 78, 1853–1860. [Google Scholar] [CrossRef]
- Huang, J.; Pan, H.; Wang, J.; Wang, T.; Huo, X.; Ma, Y.; Lu, Z.; Sun, B.; Jiang, H. Unfolded protein response in colorectal cancer. Cell Biosci. 2021, 11, 26. [Google Scholar] [CrossRef]
- Walczak, A.; Gradzik, K.; Kabzinski, J.; Przybylowska-Sygut, K.; Majsterek, I. The Role of the ER-Induced UPR Pathway and the Efficacy of Its Inhibitors and Inducers in the Inhibition of Tumor Progression. Oxid. Med. Cell. Longev. 2019, 2019, 5729710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, T.; Yoshimatsu, K.; Watanabe, K.; Yokomizo, H.; Otani, T.; Matsumoto, A.; Osawa, G.; Onda, M.; Ogawa, K. Overexpression of human X-box binding protein 1 (XBP-1) in colorectal adenomas and adenocarcinomas. Anticancer Res. 2007, 27, 127–131. [Google Scholar] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benedetti, R.; Gilardini Montani, M.S.; Romeo, M.A.; Arena, A.; Santarelli, R.; D’Orazi, G.; Cirone, M. Role of UPR Sensor Activation in Cell Death–Survival Decision of Colon Cancer Cells Stressed by DPE Treatment. Biomedicines 2021, 9, 1262. https://doi.org/10.3390/biomedicines9091262
Benedetti R, Gilardini Montani MS, Romeo MA, Arena A, Santarelli R, D’Orazi G, Cirone M. Role of UPR Sensor Activation in Cell Death–Survival Decision of Colon Cancer Cells Stressed by DPE Treatment. Biomedicines. 2021; 9(9):1262. https://doi.org/10.3390/biomedicines9091262
Chicago/Turabian StyleBenedetti, Rossella, Maria Saveria Gilardini Montani, Maria Anele Romeo, Andrea Arena, Roberta Santarelli, Gabriella D’Orazi, and Mara Cirone. 2021. "Role of UPR Sensor Activation in Cell Death–Survival Decision of Colon Cancer Cells Stressed by DPE Treatment" Biomedicines 9, no. 9: 1262. https://doi.org/10.3390/biomedicines9091262