
Fine Tuning of Cholinesterase and Glutathione-S-Transferase Activities by Organoruthenium(II) Complexes

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Synthesis
2.3. Enzyme Inhibition Assays
2.3.1. Cholinesterase Inhibition Assay
2.3.2. Glutathione S-Transferase Inhibition Assay
3. Results and Discussion
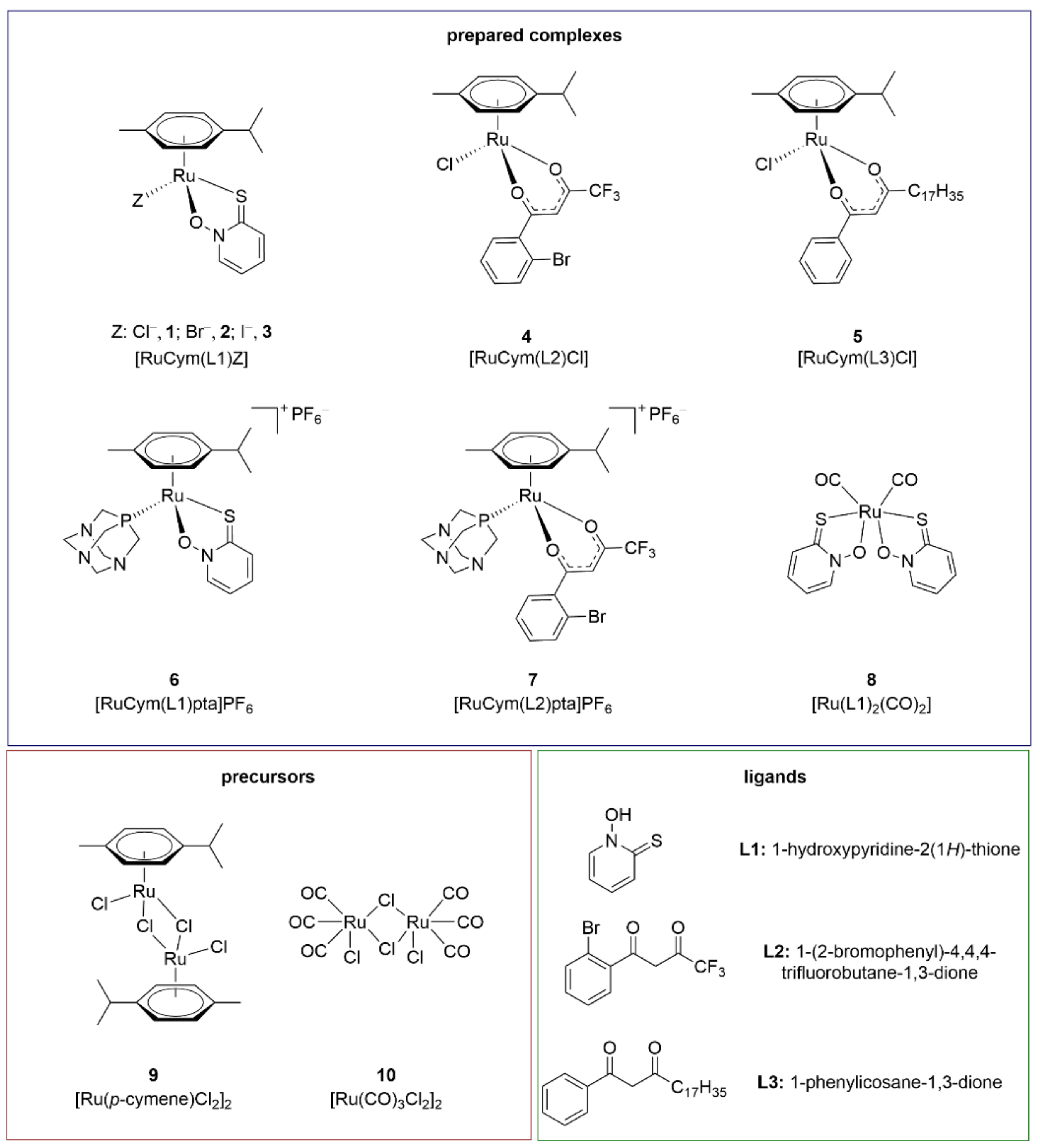
3.1. Tested Compounds and Synthesis of Organoruthenium Complexes
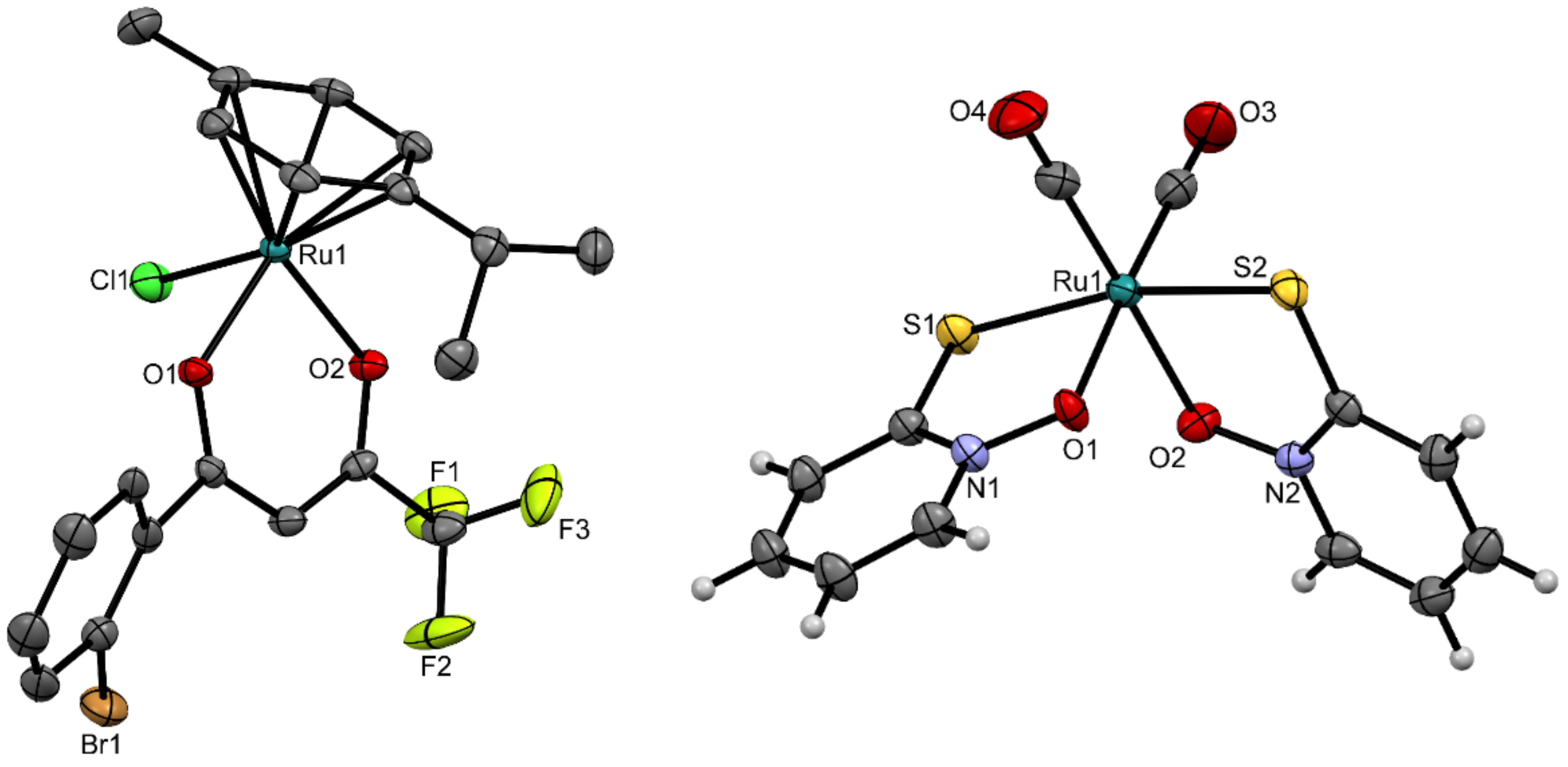
3.2. Crystal Structures
3.3. Inhibition of Cholinesterases and GSTs by the Ruthenium-Based Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clarke, M.J. Ruthenium metallopharmaceuticals. Coord. Chem. Rev. 2003, 232, 69–93. [Google Scholar] [CrossRef]
- Barry, N.P.E.; Sadler, P.J. 100 years of metal coordination chemistry: From Alfred Werner to anticancer metallodrugs. Pure Appl. Chem. 2014, 86, 1897–1910. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.J.; Sadler, P.J. Medicinal inorganic chemistry. Adv. Inorg. Chem. 1999, 49, 183–306. [Google Scholar] [CrossRef]
- Yan, Y.K.; Melchart, M.; Habtemariam, A.; Sadler, P.J. Organometallic chemistry, biology and medicine: Ruthenium arene anticancer complexes. Chem. Commun. 2005, 4764–4776. [Google Scholar] [CrossRef] [PubMed]
- Brabec, V.; Kasparkova, J. Ruthenium coordination compounds of biological and biomedical significance. DNA binding agents. Coord. Chem. Rev. 2018, 376, 75–94. [Google Scholar] [CrossRef]
- Casini, A.; Gabbiani, C.; Sorrentino, F.; Rigobello, M.P.; Bindoli, A.; Geldbach, T.J.; Marrone, A.; Re, N.; Hartinger, C.G.; Dyson, P.J.; et al. Emerging protein targets for anticancer metallodrugs: Inhibition of thioredoxin reductase and cathepsin B by antitumor ruthenium(II)-arene compounds. J. Med. Chem. 2008, 51, 6773–6781. [Google Scholar] [CrossRef]
- Che, C.M.; Siu, F.M. Metal complexes in medicine with a focus on enzyme inhibition. Curr. Opin. Chem. Biol. 2010, 14, 255–261. [Google Scholar] [CrossRef]
- Kljun, J.; Anko, M.; Traven, K.; Sinreih, M.; Pavlič, R.; Peršič, Š.; Ude, Ž.; Codina, E.E.; Stojan, J.; Lanišnik Rižner, T.; et al. Pyrithione-based ruthenium complexes as inhibitors of aldo-keto reductase 1C enzymes and anticancer agents. Dalton Trans. 2016, 45, 11791–11800. [Google Scholar] [CrossRef] [Green Version]
- Mitrovič, A.; Kljun, J.; Sosič, I.; Uršič, M.; Meden, A.; Gobec, S.; Kos, J.; Turel, I. Organoruthenated nitroxoline derivatives impair tumor cell invasion through inhibition of cathepsin B activity. Inorg. Chem. 2019, 58, 12334–12347. [Google Scholar] [CrossRef] [Green Version]
- Sundaraneedi, M.K.; Tedla, B.A.; Eichenberger, R.M.; Becker, L.; Pickering, D.; Smout, M.J.; Rajan, S.; Wangchuk, P.; Keene, F.R.; Loukas, A.; et al. Polypyridylruthenium(II) complexes exert anti-schistosome activity and inhibit parasite acetylcholinesterases. PloS Neglrcted Trop. Dis. 2017, 11, e0006134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, N.A.; Bhat, S.S.; Kumbhar, A.S.; Sonawane, U.B.; Jani, V.; Joshi, R.R.; Ramteke, S.N.; Kulkarni, P.P.; Joshi, B. Ruthenium(II) polypyridyl complex as inhibitor of acetylcholinesterase and Aβ aggregation. Eur. J. Med. Chem. 2014, 75, 375–381. [Google Scholar] [CrossRef]
- Ristovski, S.; Uzelac, M.; Kljun, J.; Lipec, T.; Uršič, M.; Jokhadar, S.Z.; Žužek, M.C.; Trobec, T.; Frangež, R.; Sepčić, K.; et al. Organoruthenium prodrugs as a new class of cholinesterase and glutathione-S-transferase inhibitors. ChemMedChem 2018, 13, 2166–2176. [Google Scholar] [CrossRef] [PubMed]
- Trobec, T.; Žužek, M.C.; Sepčić, K.; Kladnik, J.; Kljun, J.; Turel, I.; Benoit, E.; Frangež, R. Structural and functional characterization of an organometallic ruthenium complex as a potential myorelaxant drug. Biomed. Pharmacother. 2020, 127, 110161. [Google Scholar] [CrossRef]
- Colović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [Green Version]
- Pohanka, M. Cholinesterases, a target of pharmacology and toxicology. Biomed. Pap. Med Fac. Palacky Univ. Olomouc 2011, 155, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, G.; Greig, N.H.; Khan, J.A.; Kamal, M.A. Status of acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1432–1439. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.K.; Lakshmi, K.K.; Krishna, K.V.; Agrawal, M.; Singhvi, G.; Saha, R.N.; Saraf, S.; Saraf, S.; Shukla, R.; Alexander, A. Insulin mediated novel therapies for the treatment of Alzheimer’s disease. Life Sci. 2020, 249, 117540. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Sen, N. Tauopathy: A common mechanism for neurodegeneration and brain aging. Mech. Ageing Dev. 2019, 178, 72–79. [Google Scholar] [CrossRef]
- Volpato, D.; Holzgrabe, U. Designing hybrids targeting the cholinergic system by modulating the muscarinic and nicotinic receptors: A concept to treat Alzheimer’s disease. Molecules 2018, 23, 3230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, N.A.; Singh, S.B.; Kumbhar, A.S.; Ranade, D.S.; Walke, G.R.; Kulkarni, P.P.; Jani, V.; Sonavane, U.B.; Joshi, R.R.; Rapole, S. Acetylcholinesterase and Aβ aggregation inhibition by heterometallic ruthenium(II)-platinum(II) polypyridyl complexes. Inorg. Chem. 2018, 57, 7524–7535. [Google Scholar] [CrossRef]
- Dos Santos Picanco, L.C.; Ozela, P.F.; de Fatima de Brito Brito, M.; Pinheiro, A.A.; Padilha, E.C.; Braga, F.S.; de Paula da Silva, C.H.T.; Dos Santos, C.B.R.; Rosa, J.M.C.; da Silva Hage-Melim, L.I. Alzheimer’s disease: A review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Curr. Med. Chem. 2018, 25, 3141–3159. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yang, H.Y.; Chen, Y.; Sun, H.P. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 132, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef]
- Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Matic, M.; Coric, V.; Djukic, T.; Radic, T.; Simic, T. Glutathione transferases: Potential targets to overcome chemoresistance in solid tumors. Int. J. Mol. Sci. 2018, 19, 3785. [Google Scholar] [CrossRef] [Green Version]
- Di Pietro, G.; Magno, L.A.; Rios-Santos, F. Glutathione S-transferases: An overview in cancer research. Expert Opin. Drug Metab. Toxicol. 2010, 6, 153–170. [Google Scholar] [CrossRef]
- Aliende, C.; Perez-Manrique, M.; Jalon, F.A.; Manzano, B.R.; Rodriguez, A.M.; Cuevas, J.V.; Espino, G.; Martinez, M.A.; Massaguer, A.; Gonzalez-Bartulos, M.; et al. Preparation of new half sandwich ruthenium arene complexes with aminophosphines as potential chemotherapeutics. J. Inorg. Biochem. 2012, 117, 171–188. [Google Scholar] [CrossRef]
- Kljun, J.; Turel, I. β-Diketones as scaffolds for anticancer drug design—From organic building blocks to natural products and metallodrug components. Eur. J. Inorg. Chem. 2017, 1655–1666. [Google Scholar] [CrossRef] [Green Version]
- Seršen, S.; Kljun, J.; Kryeziu, K.; Panchuk, R.; Alte, B.; Korner, W.; Heffeter, P.; Berger, W.; Turel, I. Structure-related mode-of-action differences of anticancer organoruthenium complexes with β-diketonates. J. Med. Chem. 2015, 58, 3984–3996. [Google Scholar] [CrossRef]
- Vock, C.A.; Renfrew, A.K.; Scopelliti, R.; Juillerat-Jeanneret, L.; Dyson, P.J. Influence of the diketonato ligand on the cytotoxicities of [Ru(η6-p-cymene)-(R2acac)(PTA)]+ complexes (PTA = 1,3,5-triaza-7-phosphaadamantane). Eur. J. Inorg. Chem. 2008, 2008, 1661–1671. [Google Scholar] [CrossRef]
- Kladnik, J.; Coverdale, J.P.C.; Kljun, J.; Burmeister, H.; Lippman, P.; Ellis, F.G.; Jones, A.M.; Ott, I.; Romero-Canelón, I.; Turel, I. Organoruthenium complexes with benzo-fused pyrithiones overcome platinum resistance in ovarian cancer cells. Cancers 2021, 13, 2493. [Google Scholar] [CrossRef]
- Kladnik, J.; Kljun, J.; Burmeister, H.; Ott, I.; Romero-Canelón, I.; Turel, I. Towards Identification of essential structural elements of organoruthenium(II)-pyrithionato complexes for anticancer activity. Chem. Eur. J. 2019, 25, 14169–14182. [Google Scholar] [CrossRef]
- Kladnik, J.; Ristovski, S.; Kljun, J.; Defant, A.; Mancini, I.; Sepčić, K.; Turel, I. Structural isomerism and enhanced lipophilicity of pyrithione ligands of organoruthenium(II) complexes increase inhibition on AChE and BuChE. Int. J. Mol. Sci. 2020, 21, 5628. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, M.; Kluska, M.; Wysokinski, D.; Wozniak, K. DNA damage and antioxidant properties of CORM-2 in normal and cancer cells. Sci. Rep. 2020, 10, 12200. [Google Scholar] [CrossRef]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef]
- Daigle, D.J.; Decuir, T.J.; Robertson, J.B.; Darensbourg, D.J. 1,3,5–Triaz–7–phosphatricyclo[3.3.1.13,7]decane and derivatives. In Inorganic Syntheses; Darensbourg, M.Y., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1998; Volume 32, pp. 40–45. [Google Scholar]
- Willcott, M.R. MestRe Nova. J. Am. Chem. Soc. 2009, 131, 13180. [Google Scholar] [CrossRef]
- Agilent. CrysAlis PRO; Agilent Technologies Ltd.: Yarnton, UK, 2014. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Seršen, S.; Kljun, J.; Požgan, F.; Štefane, B.; Turel, I. Novel organoruthenium(II) β-diketonates as catalysts for orthoarylation via C–H activation. Organometallics 2013, 32, 609–616. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Olsen, E.K.; Hansen, E.; Moodie, L.W.K.; Isaksson, J.; Sepčić, K.; Cergolj, M.; Svenson, J.; Andersen, J.H. Marine AChE inhibitors isolated from Geodia barretti: Natural compounds and their synthetic analogs. Org. Biomol. Chem. 2016, 14, 1629–1640. [Google Scholar] [CrossRef]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [CrossRef]
- Habtemariam, A.; Melchart, M.; Fernández, R.; Parsons, S.; Oswald, I.D.H.; Parkin, A.; Fabbiani, F.P.A.; Davidson, J.E.; Dawson, A.; Aird, R.E.; et al. Structure-activity relationships for cytotoxic ruthenium(II) arene complexes containing N,N-, N,O-, and O,O-chelating ligands. J. Med. Chem. 2006, 49, 6858–6868. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Soukup, O.; Winder, M.; Killi, U.K.; Wsol, V.; Jun, D.; Kuca, K.; Tobin, G. Acetylcholinesterase inhibitors and drugs acting on muscarinic receptors- potential crosstalk of cholinergic mechanisms during pharmacological treatment. Curr. Neuropharmacol. 2017, 15, 637–653. [Google Scholar] [CrossRef] [Green Version]
- Lanni, C.; Masi, M.; Racchi, M.; Govoni, S. Cancer and Alzheimer’s disease inverse relationship: An age-associated diverging derailment of shared pathways. Mol. Psychiatr. 2021, 26, 280–295. [Google Scholar] [CrossRef]
- Dvir, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D structure to function. Chem. Biol. Interact. 2010, 187, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alatrash, N.; Narh, E.S.; Yadav, A.; Kim, M.J.; Janaratne, T.; Gabriel, J.; MacDonnell, F.M. Synthesis, DNA cleavage activity, cytotoxicity, acetylcholinesterase inhibition, and acute murine toxicity of redox-active ruthenium(II) polypyridyl complexes. ChemMedChem 2017, 12, 1055–1069. [Google Scholar] [CrossRef]
- Cardoso, C.R.; de Aguiar, I.; Camilo, M.R.; Lima, M.V.; Ito, A.S.; Baptista, M.S.; Pavani, C.; Venancio, T.; Carlos, R.M. Synthesis, spectroscopic characterization, photochemical and photophysical properties and biological activities of ruthenium complexes with mono- and bi-dentate histamine ligand. Dalton Trans. 2012, 41, 6726–6734. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, S.P.; Li, S.; Korn, R.; Xie, X.; Meggers, E. Solid-phase synthesis of tris-heteroleptic ruthenium(II) complexes and application to acetylcholinesterase inhibition. Inorg. Chem. 2008, 47, 5030–5032. [Google Scholar] [CrossRef]
- Silva, D.E.; Cali, M.P.; Pazin, W.M.; Carlos-Lima, E.; Salles Trevisan, M.T.; Venancio, T.; Arcisio-Miranda, M.; Ito, A.S.; Carlos, R.M. Luminescent Ru(II) phenanthroline complexes as a probe for real-time imaging of Aβ self-aggregation and therapeutic applications in Alzheimer’s Disease. J. Med. Chem. 2016, 59, 9215–9227. [Google Scholar] [CrossRef]
- Kubanik, M.; Holtkamp, H.; Söhnel, T.; Jamieson, S.M.F.; Hartinger, C.G. Impact of the halogen substitution pattern on the biological activity of organoruthenium 8-hydroxyquinoline anticancer agents. Organometallics 2015, 34, 5658–5668. [Google Scholar] [CrossRef]
- Romero-Canelón, I.; Pizarro, A.M.; Habtemariam, A.; Sadler, P.J. Contrasting cellular uptake pathways for chlorido and iodido iminopyridine ruthenium arene anticancer complexes. Metallomics 2012, 4, 1271–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Habtemariam, A.; van der Geer, E.P.L.; Fernandez, R.; Melchart, M.; Deeth, R.J.; Aird, R.; Guichard, S.; Fabbiani, F.P.A.; Lozano-Casal, P.; et al. Controlling ligand substitution reactions of organometallic complexes: Tuning cancer cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18269–18274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.K. Role of carbon monoxide in neurovascular repair processing. Biomol. Ther. 2018, 26, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Hettiarachchi, N.; Dallas, M.; Al-Owais, M.; Griffiths, H.; Hooper, N.; Scragg, J.; Boyle, J.; Peers, C. Heme oxygenase-1 protects against Alzheimer’s amyloid-β(1-42)-induced toxicity via carbon monoxide production. Cell Death Dis. 2014, 5, e1569. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Enzyme Inhibition (μM) | |||||
|---|---|---|---|---|---|
| eeAChE | hrAChE | ||||
| Compound | Code | IC50 | Ki | IC50 | Ki |
| [RuCym(L1)Cl] a | 1 | 5.01 ± 0.8 | 35.0 | 25.06 ± 2.5 | 24.0 |
| [RuCym(L1)Br] | 2 | 13.14 ± 2.5 | 4.87 | 6.57 ± 4.1 | 2.49 |
| [RuCym(L1)I] | 3 | 12.90 ± 2.0 | 1.43 | 6.55 ± 4.5 | 1.94 |
| [RuCym(L2)Cl] | 4 | / | / | / | / |
| [RuCym(L3)Cl] | 5 | / | / | / | / |
| [RuCym(L1)pta]PF6 b | 6 | / | / | / | / |
| [RuCym(L2)pta]PF6 | 7 | / | / | / | / |
| [Ru(L1)2(CO)2] | 8 | / | / | / | / |
| [Ru(p-cymene)Cl2]2 a | 9 | >100 | / | >100 | / |
| [Ru(CO)3Cl2]2 | 10 | >100 | / | / | / |
| 1-Hydroxypyridine-2(1H)-thione | L1 | >100 | / | / | / |
| 1-(2-Bromophenyl)-4,4,4-trifluorobutane-1,3-dione | L2 | / | / | / | / |
| 1-Phenylicosane-1,3-dione | L3 | / | / | / | / |
| Neostigmine methylsulphate | 5.98 ± 1.0 | / | / | / | |
| Enzyme Inhibition (μM) | |||
|---|---|---|---|
| hsBChE | |||
| Compound | Code | IC50 | Ki |
| [RuCym(L1)Cl] a | 1 | 7.52 ± 1.3 | 4.0 |
| [RuCym(L1)Br] | 2 | 3.39 ± 2.3 | 0.63 |
| [RuCym(L1)I] | 3 | 3.48 ± 0.9 | 0.80 |
| [RuCym(L2)Cl] | 4 | 30.98 ± 2.1 | 6.19 |
| [RuCym(L3)Cl] | 5 | 31.99 ± 2.5 | 8.84 |
| [RuCym(L1)pta]PF6 b | 6 | 0.39 ± 0.79 | 1.1 |
| [RuCym(L2)pta]PF6 | 7 | 19.20 ± 1.5 | 9.26 |
| [Ru(L1)2(CO)2] | 8 | / | / |
| [Ru(p-cymene)Cl2]2 a | 9 | 32.70 ± 4.3 | / |
| [Ru(CO)3Cl2]2 | 10 | >100 | / |
| 1-Hydroxypyridine-2(1H)-thione | L1 | >100 | / |
| 1-(2-Bromophenyl)-4,4,4-trifluorobutane-1,3-dione | L2 | / | / |
| 1-Phenylicosane-1,3-dione | L3 | / | / |
| Neostigmine methylsulphate | 92.70 ± 2.2 | / | |
| Enzyme Inhibition (μM) | |||||
|---|---|---|---|---|---|
| hlGST | hGST | ||||
| Compound | Code | IC50 | Ki | IC50 | Ki |
| [RuCym(L1)Cl] a | 1 | 2.26 ± 0.5 | 10.0 | 45.0 ± 5.2 | / |
| [RuCym(L1)Br] | 2 | <3.39 | 0.79 | 4.64 ± 3.7 | 4.08 |
| [RuCym(L1)I] | 3 | <3.07 | 1.60 | 15.97 ± 3.0 | 8.60 |
| [RuCym(L2)Cl] | 4 | 16.11 ± 6.5 | 2.83 | >100 | / |
| [RuCym(L3)Cl] | 5 | 18.28 ± 5.3 | 1.83 | / | / |
| [RuCym(L1)pta]PF6 b | 6 | / | / | * | * |
| [RuCym(L2)pta]PF6 | 7 | / | / | / | / |
| [Ru(L1)2(CO)2] | 8 | <3.66 | 0.85 | 16.61 ± 1.4 | 9.65 |
| [Ru(p-cymene)Cl2]2 a | 9 | >100 | / | >100 | / |
| [Ru(CO)3Cl2]2 | 10 | 9.76 ± 0.4 | 2.93 | 97.65 ± 1.0 | / |
| 1-Hydroxypyridine-2(1H)-thione | L1 | / | / | / | / |
| 1-(2-Bromophenyl)-4,4,4-trifluorobutane-1,3-dione | L2 | / | / | / | / |
| 1-Phenylicosane-1,3-dione | L3 | / | / | / | / |
| Neostigmine methylsulphate | / | / | / | / | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trobec, T.; Sepčić, K.; Žužek, M.C.; Kladnik, J.; Podjed, N.; Cardoso Páscoa, C.; Turel, I.; Frangež, R. Fine Tuning of Cholinesterase and Glutathione-S-Transferase Activities by Organoruthenium(II) Complexes. Biomedicines 2021, 9, 1243. https://doi.org/10.3390/biomedicines9091243
Trobec T, Sepčić K, Žužek MC, Kladnik J, Podjed N, Cardoso Páscoa C, Turel I, Frangež R. Fine Tuning of Cholinesterase and Glutathione-S-Transferase Activities by Organoruthenium(II) Complexes. Biomedicines. 2021; 9(9):1243. https://doi.org/10.3390/biomedicines9091243
Chicago/Turabian StyleTrobec, Tomaž, Kristina Sepčić, Monika Cecilija Žužek, Jerneja Kladnik, Nina Podjed, Catarina Cardoso Páscoa, Iztok Turel, and Robert Frangež. 2021. "Fine Tuning of Cholinesterase and Glutathione-S-Transferase Activities by Organoruthenium(II) Complexes" Biomedicines 9, no. 9: 1243. https://doi.org/10.3390/biomedicines9091243