
Role of Calcium Homeostasis in Modulating EMT in Cancer


Abstract
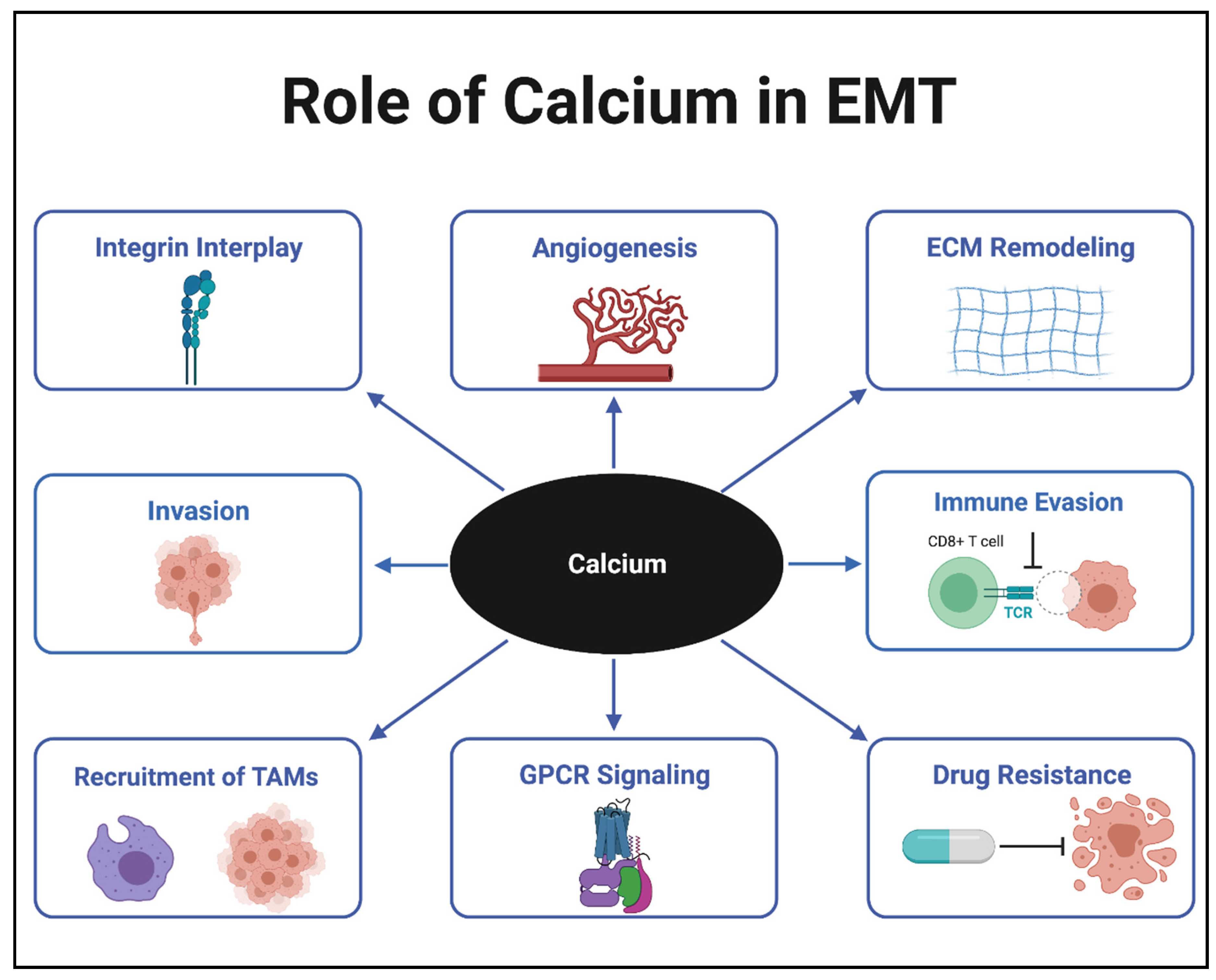
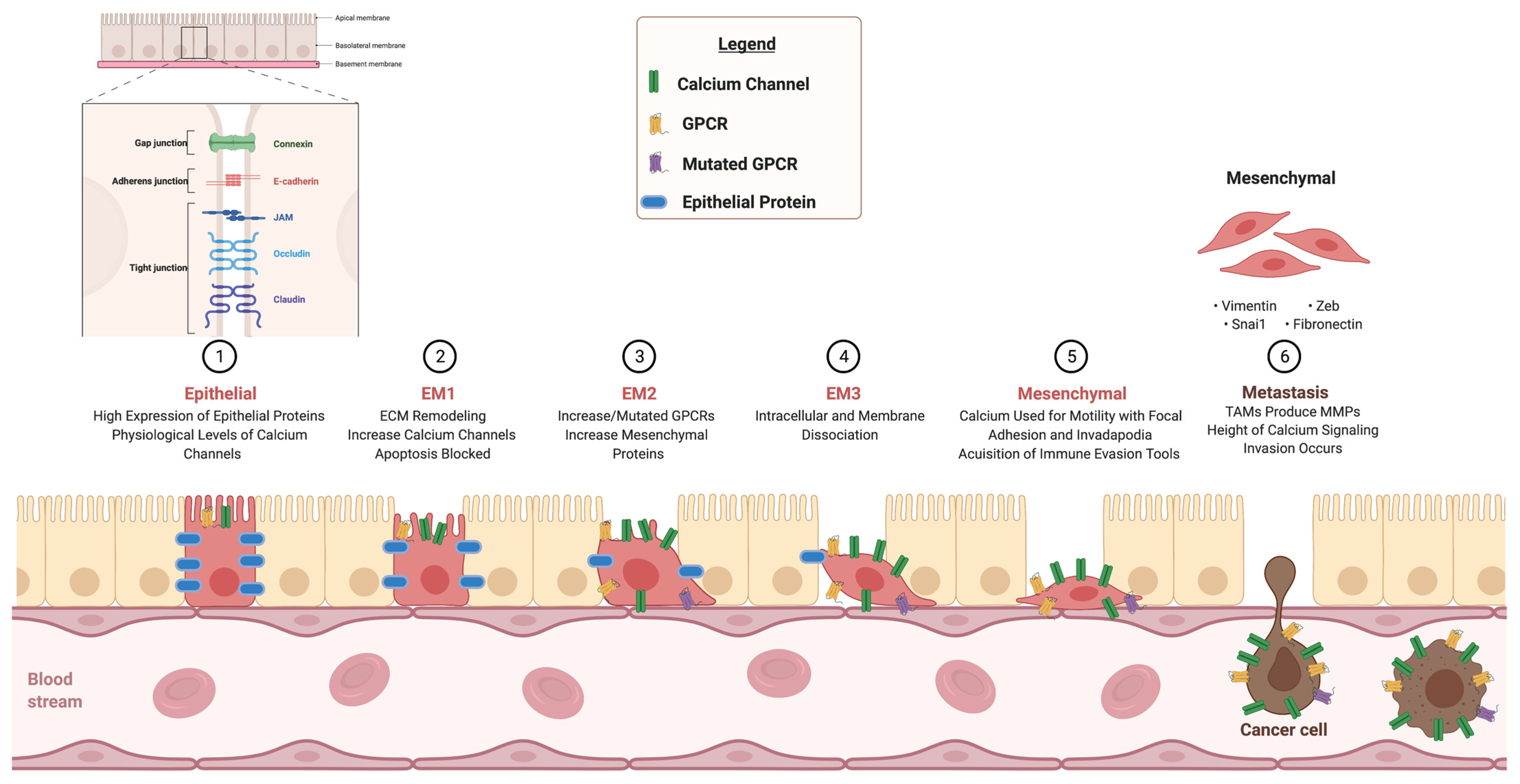
:1. Introduction
2. Calcium Channels
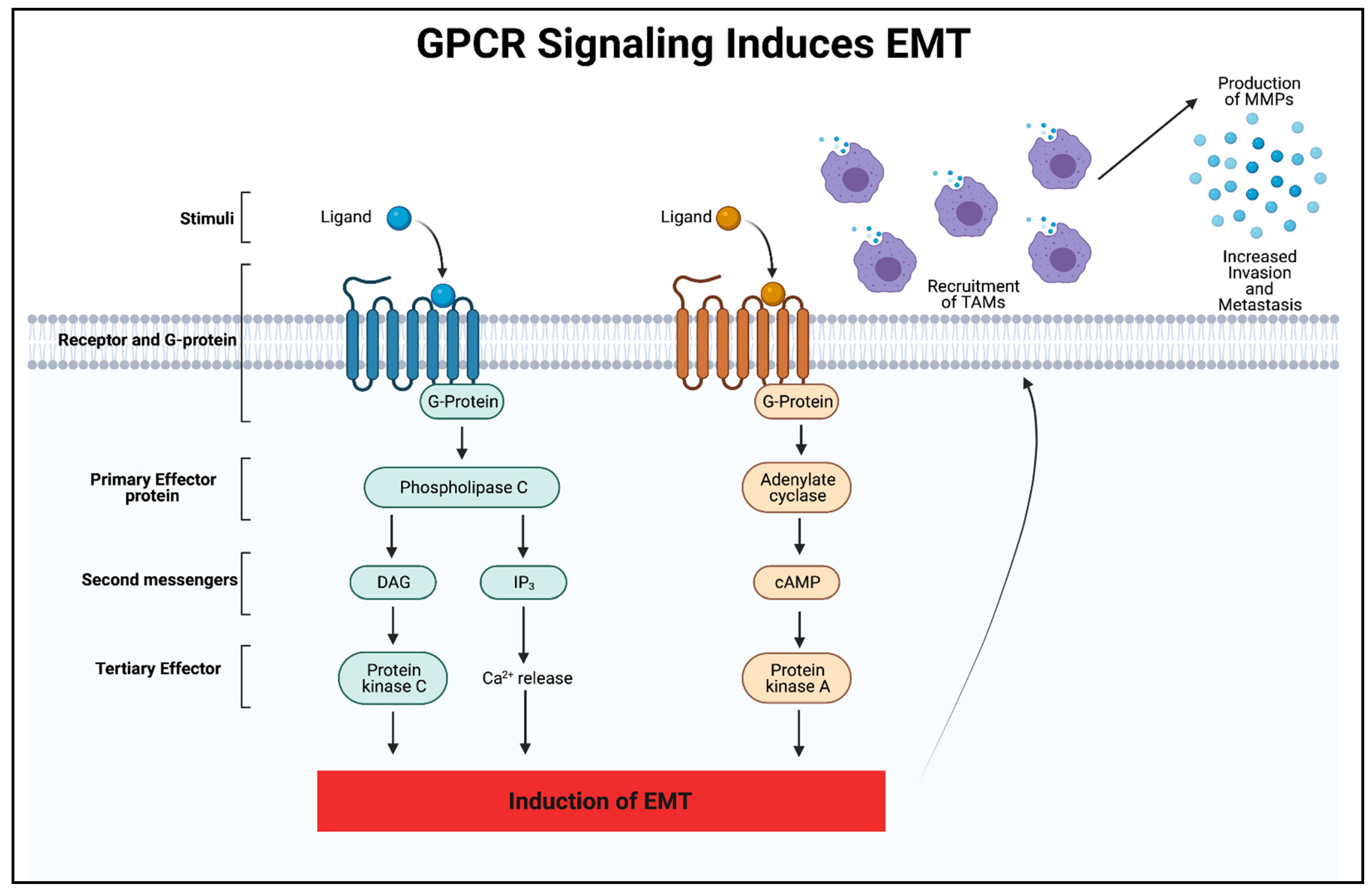
3. GPCR Signaling
4. Integrins
5. Calcium in Immune Evasion and Drug Resistance
6. Combination Therapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Terrié, E.; Coronas, V.; Constantin, B. Role of the calcium toolkit in cancer stem cells. Cell Calcium 2019, 80, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Immler, R.; Simon, S.I.; Sperandio, M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur. J. Clin. Investig. 2018, 48, e12964. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ sensing: Its role in calcium homeostasis and signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, C.M.; Peacock, M. Calcium. Adv. Nutr. 2019, 10, 546–548. [Google Scholar] [CrossRef]
- Esbrit, P.; Alcaraz, M.J. Current perspectives on parathyroid hormone (PTH) and PTH-related protein (PTHrP) as bone anabolic therapies. Biochem. Pharmacol. 2013, 85, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Xu, Y.; Li, L.-X. The coexistence of hypercalcemia, osteoporosis and thymic enlargement in graves’ disease: A case report. BMC Endocr. Disord. 2020, 20, 97. [Google Scholar] [CrossRef]
- Stewart, T.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 2502–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, L.F.; Gao, J.H.; Li, L.; Jiang, P.; Lv, X.; Yu, L.X.; Yang, J.; Li, R.T.; Liu, B.R. Clinical significance of epithelial–mesenchymal transition-related molecules in lung adenocarcinoma. Curr. Oncol. 2019, 26, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Elzamly, S.; Badri, N.; Padilla, O.; Dwivedi, A.K.; Alvarado, L.A.; Hamilton, M.; Diab, N.; Rock, C.; Elfar, A.; Teleb, M.; et al. Epithelial-mesenchymal transition markers in breast cancer and pathological responseafter neoadjuvant chemotherapy. Breast Cancer Basic Clin. Res. 2018, 12, 117822341878807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyuno, D.; Yamaguchi, H.; Ito, T.; Kono, T.; Kimura, Y.; Imamura, M.; Konno, T.; Hirata, K.; Sawada, N.; Kojima, T. Targeting tight junctions during epithelial to mesenchymal transition in human pancreatic cancer. World J. Gastroenterol. 2014, 20, 10813–10824. [Google Scholar] [CrossRef]
- Cahalan, M.D. How to STIMulate calcium channels. Science 2010, 330, 43–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, K.; Liskova, A.; Kubatka, P.; Büsselberg, D. Calcium entry through TRPV1: A potential target for the regulation of proliferation and apoptosis in cancerous and healthy cells. Int. J. Mol. Sci. 2020, 21, 4177. [Google Scholar] [CrossRef]
- Collins, H.E.; Zhu-Mauldin, X.; Marchase, R.B.; Chatham, J.C. STIM1/Orai1-mediated SOCE: Current perspectives and potential roles in cardiac function and pathology. Am. J. Physiol. Circ. Physiol. 2013, 305, H446–H458. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Bon, R.; Beech, D.J. In pursuit of small molecule chemistry for calcium-permeable non-selective TRPC channels—Mirage or pot of gold? Br. J. Pharmacol. 2013, 170, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Worley, P.F.; Zeng, W.; Huang, G.N.; Yuan, J.P.; Kim, J.Y.; Lee, M.G.; Muallem, S. TRPC channels as STIM1-regulated store-operated channels. Cell Calcium 2007, 42, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.T.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Contribution of TRPC1 and Orai1 to Ca2+ entry activated by store depletion. In Advances in Experimental Medicine and Biology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2011; Volume 704, pp. 435–449. [Google Scholar]
- Asghar, M.Y.; Törnquist, K. Transient receptor potential canonical (TRPC) channels as modulators of migration and invasion. Int. J. Mol. Sci. 2020, 21, 1739. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Xu, H. High expression of TRPM8 predicts poor prognosis in patients with osteosarcoma. Oncol. Lett. 2016, 12, 1373–1379. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.-S.; Wen, J.; Yang, F.; Cai, X.-L.; Yang, H.; Luo, K.-J.; Liu, Q.W.; Hu, R.-G.; Xie, X.; Huang, Q.-Y.; et al. High expression of transient potential receptor C6 correlated with poor prognosis in patients with esophageal squamous cell carcinoma. Med. Oncol. 2013, 30, 1–8. [Google Scholar] [CrossRef]
- Karacicek, B.; Erac, Y.; Tosun, M. Functional consequences of enhanced expression of STIM1 and Orai1 in Huh-7 hepatocellular carcinoma tumor-initiating cells. BMC Cancer 2019, 19, 751. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Z.-Y.; Zhong, L.-X.; Feng, M.; Wang, J.-F.; Liu, D.-B.; Xiong, J.-P. Over-expression of Orai1 mediates cell proliferation and associates with poor prognosis in human non-small cell lung carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 5080–5088. [Google Scholar] [PubMed]
- Wang, Z. Transactivation of epidermal growth factor receptor by G protein-coupled receptors: Recent progress, challenges and future research. Int. J. Mol. Sci. 2016, 17, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.J.; Sümbül, U.; Graybuck, L.T.; Collman, F.; Seshamani, S.; Gala, R.; Gliko, O.; Elabbady, L.; Miller, J.A.; Bakken, T.E.; et al. Single-cell transcriptomic evidence for dense intracortical neuropeptide networks. eLife 2019, 8, 47889. [Google Scholar] [CrossRef]
- Lämmermann, T.; Kastenmüller, W. Concepts of GPCR-controlled navigation in the immune system. Immunol. Rev. 2019, 289, 205–231. [Google Scholar] [CrossRef] [Green Version]
- O’Hayre, M.; Degese, M.S.; Gutkind, J.S. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr. Opin. Cell Biol. 2014, 27, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Xie, A.X.; Madayag, A.; Minton, S.K.; McCarthy, K.D.; Malykhina, A.P. Sensory satellite glial Gq-GPCR activation alleviates inflammatory pain via peripheral adenosine 1 receptor activation. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Kimura, T.; Pydi, S.P.; Pham, J.; Tanaka, N. Metabolic functions of G protein-coupled receptors in hepatocytes—Potential applications for diabetes and NAFLD. Biomolecules 2020, 10, 1445. [Google Scholar] [CrossRef]
- Ahmad, R.; Dalziel, J.E. G protein-coupled receptors in taste physiology and pharmacology. Front. Pharmacol. 2020, 11, 587664. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-Y.; Wu, J.-J.; Peng, W.-T.; Sun, J.-C.; Wei, W. The role of G protein-coupled receptor kinases in the pathology of malignant tumors. Acta Pharmacol. Sin. 2018, 39, 1699–1705. [Google Scholar] [CrossRef] [Green Version]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef]
- Rohacs, T. Regulation of transient receptor potential channels by the phospholipase C pathway. Adv. Biol. Regul. 2013, 53, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Thakur, D.P.; Tian, J.; Jeon, J.; Xiong, J.; Huang, Y.; Flockerzi, V.; Zhu, M.X. Critical roles of Gi/o proteins and phospholipase C-δ1 in the activation of receptor-operated TRPC4 channels. Proc. Natl. Acad. Sci. USA 2016, 113, 1092–1097. [Google Scholar] [CrossRef] [Green Version]
- Sriram, K.; Moyung, K.; Corriden, R.; Carter, H.; Insel, P.A. GPCRs show widespread differential mRNA expression and frequent mutation and copy number variation in solid tumors. PLoS Biol. 2019, 17, e3000434. [Google Scholar] [CrossRef] [Green Version]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G protein-coupled receptors in cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef] [Green Version]
- Kumari, N.; Reabroi, S.; North, B.J. Unraveling the molecular nexus between GPCRs, ERS, and EMT. Mediat. Inflamm. 2021, 2021, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Viloria-Petit, A.M.; Wrana, J.L. The TGFβ-Par6 polarity pathway: Linking the par complex to EMT and breast cancer progression. Cell Cycle 2010, 9, 623–624. [Google Scholar] [CrossRef]
- Di Roberto, R.B.; Chang, B.; Peisajovich, S.G. The directed evolution of ligand specificity in a GPCR and the unequal contributions of efficacy and affinity. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jespers, W.; Bongers, B.J.; Jansen, M.C.H.; Stangenberger, C.M.; Dilweg, M.; Gutiérrez-de-Terán, H.; Ijzerman, A.P.; Heitman, L.H.; van Westen, G.J. Characterization of cancer-related somatic mutations in the adenosine A2B receptor. Eur. J. Pharmacol. 2020, 880, 173126. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Tan, P.; Rodriguez, M.; He, L.; Tan, K.; Zeng, L.; Siwko, S.; Liu, M. Leucine-rich repeat-containing G protein-coupled receptor 4 (Lgr4) is necessary for prostate cancer metastasis via epithelial-mesenchymal transition. J. Biol. Chem. 2017, 292, 15525–15537. [Google Scholar] [CrossRef] [Green Version]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Tharmalingam, S.; Hampson, D.R. The calcium-sensing receptor and integrins in cellular differentiation and migration. Front. Physiol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Badaoui, M.; Mimsy-Julienne, C.; Saby, C.; van Gulick, L.; Peretti, M.; Jeannesson, P.; Morjani, H.; Ouadid-Ahidouch, H. Collagen type 1 promotes survival of human breast cancer cells by overexpressing Kv10. 1 potassium and Orai1 calcium channels through DDR1-dependent pathway. Oncotarget 2017, 9, 24653–24671. [Google Scholar] [CrossRef]
- Tiwari, S.; Askari, J.A.; Humphries, M.; Bulleid, N.J. Divalent cations regulate the folding and activation status of integrins during their intracellular trafficking. J. Cell Sci. 2011, 124, 1672–1680. [Google Scholar] [CrossRef] [Green Version]
- Kirchhofer, D.; Grzesiak, J.; Pierschbacher, M.D. Calcium as a potential physiological regulator of integrin-mediated cell adhesion. J. Biol. Chem. 1991, 266, 4471–4477. [Google Scholar] [CrossRef]
- Jacquemet, G.; Baghirov, H.; Georgiadou, M.; Sihto, H.; Peuhu, E.; Cettour-Janet, P.; He, T.; Perälä, M.; Kronqvist, P.; Joensuu, H.; et al. L-type calcium channels regulate filopodia stability and cancer cell invasion downstream of integrin signalling. Nat. Commun. 2016, 7, 13297. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhan, H. Communication between EMT and PD-L1 signaling: New insights into tumor immune evasion. Cancer Lett. 2020, 468, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.-M.; Xia, W.; Hsu, Y.-H.; Chan, L.-C.; Yu, W.-H.; Cha, J.-H.; Chen, C.-T.; Liao, H.-W.; Kuo, C.-W.; Khoo, K.-H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Kumar, S.; Davra, V.; Obr, A.E.; Geng, K.; Wood, T.; de Lorenzo, M.S.; Birge, R.B. Crk adaptor protein promotes PD-L1 expression, EMT and immune evasion in a murine model of triple-negative breast cancer. Oncoimmunology 2017, 7, e1376155. [Google Scholar] [CrossRef] [PubMed]
- Datar, I.; Schalper, K.A. Epithelial-mesenchymal transition and immune evasion during lung cancer progression: The chicken or the egg? Clin. Cancer Res. 2016, 22, 3422–3424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.-P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Chen, L.; Liu, L.; Niu, X. EMT-mediated acquired EGFR-TKI resistance in NSCLC: Mechanisms and strategies. Front. Oncol. 2019, 9, 1044. [Google Scholar] [CrossRef] [Green Version]
- Vijay, G.V.; Zhao, N.; Hollander, P.D.; Toneff, M.J.; Joseph, R.; Pietila, M.; Taube, J.H.; Sarkar, T.R.; Ramirez-Pena, E.; Werden, S.J.; et al. GSK3β regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res. 2019, 21, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, C.; Wang, Y. The importance of epithelial-mesenchymal transition and autophagy in cancer drug resistance. Cancer Drug Resist. 2019, 3, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, L.; Lalier, L.; Salaud, C.; Heymann, D.; Cartron, P.F.; Vallette, F.M. Drug resistance in glioblastoma: Are persisters the key to therapy? Cancer Drug Resist. 2020. [Google Scholar] [CrossRef]
- Han, R.-F.; Ji, X.; Dong, X.-G.; Xiao, R.-J.; Liu, Y.-P.; Xiong, J.; Zhang, Q.-P. An epigenetic mechanism underlying doxorubicin induced EMT in the human BGC-823 gastric cancer cell. Asian Pac. J. Cancer Prev. 2014, 15, 4271–4274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Tong, D.; Liu, G.; Xu, J.; Do, K.; Geary, K.; Zhang, D.; Zhang, J.; Zhang, Y.; Li, Y.; et al. Metformin reverses prostate cancer resistance to enzalutamide by targeting TGF-β1/STAT3 axis-regulated EMT. Cell Death Dis. 2017, 8, e3007. [Google Scholar] [CrossRef]
- Liang, Y.; Liang, Q.; Qiao, L.; Xiao, F. MicroRNAs modulate drug resistance-related mechanisms in hepatocellular carcinoma. Front. Oncol. 2020, 10, 920. [Google Scholar] [CrossRef]
- Jing, L.; Bo, W.; Yourong, F.; Tian, W.; Shixuan, W.; Mingfu, W. Sema4C mediates EMT inducing chemotherapeutic resistance of miR-31-3p in cervical cancer cells. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Kurimoto, R.; Iwasawa, S.; Ebata, T.; Ishiwata, T.; Sekine, I.; Tada, Y.; Tatsumi, K.; Koide, S.; Iwama, A.; Takiguchi, Y. Drug resistance originating from a TGF-β/FGF-2-driven epithelial-to-mesenchymal transition and its reversion in human lung adenocarcinoma cell lines harboring an EGFR mutation. Int. J. Oncol. 2016, 48, 1825–1836. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wu, C.; Liang, H.; Zhao, Y.; Lin, C.; Zhang, X.; Ye, C. Knockdown of TWIST enhances the cytotoxicity of chemotherapeutic drugs in doxorubicin-resistant HepG2 cells by suppressing MDR1 and EMT. Int. J. Oncol. 2018, 53, 1763–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vig, M.; Kinet, J.-P. Calcium signaling in immune cells. Nat. Immunol. 2009, 10, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Yao, Y.; Gong, C.; Yu, F.; Su, S.; Chen, J.; Liu, B.; Deng, H.; Wang, F.; Lin, L.; et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 2011, 19, 541–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Friedmann, K.S.; Lyrmann, H.; Zhou, Y.; Schoppmeyer, R.; Knörck, A.; Mang, S.; Hoxha, C.; Angenendt, A.; Backes, C.S.; et al. A calcium optimum for cytotoxic T lymphocyte and natural killer cell cytotoxicity. J. Physiol. 2018, 596, 2681–2698. [Google Scholar] [CrossRef] [Green Version]
- Scimeca, M.; Urbano, N.; Rita, B.; Mapelli, S.N.; Catapano, C.V.; Carbone, G.M.; Ciuffa, S.; Tavolozza, M.; Schillaci, O.; Mauriello, A.; et al. Prostate osteoblast-like cells: A reliable prognostic marker of bone metastasis in prostate cancer patients. Contrast Media Mol. Imaging 2018, 2018, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Scimeca, M.; Bonfiglio, R.; Menichini, E.; Albonici, L.; Urbano, N.; de Caro, M.T.; Mauriello, A.; Schillaci, O.; Gambacurta, A.; Bonanno, E. Microcalcifications drive breast cancer occurrence and development by macrophage-mediated epithelial to mesenchymal transition. Int. J. Mol. Sci. 2019, 20, 5633. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-Y.; Chen, B.-K.; Wang, Y.-S.; Tsai, Y.-T.; Chen, W.-C.; Chang, W.-C.; Hou, M.-F.; Wu, Y.-C.; Chang, W.-C. Involvement of store-operated calcium signaling in EGF-mediated COX-2 gene activation in cancer cells. Cell. Signal. 2012, 24, 162–169. [Google Scholar] [CrossRef]
- Raynal, N.J.-M.; Lee, J.T.; Wang, Y.; Beaudry, A.; Madireddi, P.; Garriga, J.; Malouf, G.; Dumont, S.N.; Dettman, E.J.; Gharibyan, V.; et al. Targeting calcium signaling induces epigenetic reactivation of tumor suppressor genes in cancer. Cancer Res. 2016, 76, 1494–1505. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Kumar, J.; Hermanson, K.; Sun, Y.; Qureshi, H.; Perley, D.; Scheidegger, A.; Singh, B.; Dhasarathy, A. The calcium channel proteins ORAI3 and STIM1 mediate TGF-β induced Snai1 expression. Oncotarget 2018, 9, 29468–29483. [Google Scholar] [CrossRef] [Green Version]
- Battista, T.; Fiorillo, A.; Chiarini, V.; Genovese, I.; Ilari, A.; Colotti, G. Roles of sorcin in drug resistance in cancer: One protein, many mechanisms, for a novel potential anticancer drug target. Cancers 2020, 12, 887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Angelis, M.L.; Francescangeli, F.; La Torre, F.; Zeuner, A. Stem cell plasticity and dormancy in the development of cancer therapy resistance. Front. Oncol. 2019, 9, 626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, C.; Prust, N.; van Doorn, S.; Reinecke, M.; Kuster, B.; en Henegouwen, P.V.B.; Lemeer, S. Adaptive resistance to EGFR-targeted therapy by calcium signaling in NSCLC cells. Mol. Cancer Res. 2018, 16, 1773–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, F.Z.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular heterogeneity in lung cancer: From mechanisms of origin to clinical implications. Int. J. Med. Sci. 2019, 16, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Turashvili, G.; Brogi, E. Tumor heterogeneity in breast cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carm, K.T.; Hoff, A.M.; Bakken, A.C.; Axcrona, U.; Axcrona, K.; Lothe, R.A.; Skotheim, R.I.; Løvf, M. Interfocal heterogeneity challenges the clinical usefulness of molecular classification of primary prostate cancer. Sci. Rep. 2019, 9, 13579. [Google Scholar] [CrossRef] [Green Version]
- Ljungberg, B.; Mehle, C.; Stenling, R.; Roos, G. Heterogeneity in renal cell carcinoma and its impact on prognosis—A flow cytometric study. Br. J. Cancer 1996, 74, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Saphire, E.O. A glimpse into immune responses evolving against Ebola virus. Nat. Med. 2019, 25, 1470–1471. [Google Scholar] [CrossRef]
- Rajan, S.; Cam, M.; Gross, A.C.; Taslim, C.; Wang, M.; Franz, E.; Roberts, R.D. Osteosarcoma tumors maintain intratumoral heterogeneity, even while adapting to environmental pressures that drive clonal selection. Cancer Biol. 2020. bioRxiv preprint. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and intertumor heterogeneity in melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef]
- Akram, M.; Iqbal, M.; Daniyal, M.; Khan, A.U. Awareness and current knowledge of breast cancer. Biol. Res. 2017, 50, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, B.K.; Deng, C.-X. Characterization of potential driver mutations involved in human breast cancer by computational approaches. Oncotarget 2017, 8, 50252–50272. [Google Scholar] [CrossRef] [Green Version]
- Makena, M.R.; Rao, R. Subtype specific targeting of calcium signaling in breast cancer. Cell Calcium 2020, 85, 102109. [Google Scholar] [CrossRef]
- Fu, S.; Hirte, H.; Welch, S.; Ilenchuk, T.T.; Lutes, T.; Rice, C.; Fields, N.; Nemet, A.; Dugourd, D.; Piha-Paul, S.; et al. First-in-human phase I study of SOR-C13, a TRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Varghese, E.; Samuel, S.M.; Sadiq, Z.; Kubatka, P.; Liskova, A.; Benacka, J.; Pazinka, P.; Kruzliak, P.; Büsselberg, D. Anti-cancer agents in proliferation and cell death: The calcium connection. Int. J. Mol. Sci. 2019, 20, 3017. [Google Scholar] [CrossRef] [Green Version]
- Bryant, J.A.; Finn, R.S.; Slamon, D.J.; Cloughesy, T.F.; Charles, A.C. EGF activates intracellular and intercellular calcium signaling by distinct pathways in tumor cells. Cancer Biol. Ther. 2004, 3, 1243–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Li, R.; Guo, H.; Zhang, W.; Xu, X.; Chen, X.; Ding, L. Dihydropyridine calcium channel blockers suppress the transcription of PD-L1 by inhibiting the activation of STAT1. Front. Pharmacol. 2021, 11, 539261. [Google Scholar] [CrossRef]
- Leung, L.; Mok, T.S.; Loong, H. Combining chemotherapy with epidermal growth factor receptor inhibition in advanced non-small cell lung cancer. Ther. Adv. Med. Oncol. 2012, 4, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Sun, W.; Zhou, Y.; Li, P.; Chen, F.; Chen, H.; Xia, D.; Xu, E.; Lai, M.; Wu, Y.; et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018, 37, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef]
- Can, G.; Akpinar, B.; Baran, Y.; Zhivotovsky, B.; Olsson, M. 5-Fluorouracil signaling through a calcium–calmodulin-dependent pathway is required for p53 activation and apoptosis in colon carcinoma cells. Oncogene 2013, 32, 4529–4538. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, M.G.; Sánchez, A.M.; Collado, B.; Malagarie-Cazenave, S.; Olea, N.; Carmena, M.J.; Prieto, J.C.; Díaz-Laviada, I. Expression of the transient receptor potential vanilloid 1 (TRPV1) in LNCaP and PC-3 prostate cancer cells and in human prostate tissue. Eur. J. Pharmacol. 2005, 515, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Lei, Y.; Li, G.; Cheng, Y.; Yang, H.; Xie, L.; Long, H.; Jiang, R. Integrative analysis of cancer driver genes in prostate adenocarcinoma. Mol. Med. Rep. 2019, 19, 2707–2715. [Google Scholar] [CrossRef]
- Lee, H.S.; Park, S.W. Systemic chemotherapy in advanced pancreatic cancer. Gut Liver 2016, 10, 340–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holdhoff, M.; Ye, X.; Supko, J.G.; Nabors, L.; Desai, A.S.; Walbert, T.; Lesser, G.J.; Read, W.L.; Lieberman, F.S.; Lodge, M.A.; et al. Timed sequential therapy of the selective T-type calcium channel blocker mibefradil and temozolomide in patients with recurrent high-grade gliomas. Neuro-Oncology 2017, 19, 845–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaev, S.I.; Santoni, F.; Garieri, M.; Makrythanasis, P.; Falconnet, E.; Guipponi, M.; Vannier, A.; Radovanovic, I.; Bena, F.S.; Forestier, F.; et al. Extrachromosomal driver mutations in glioblastoma and low-grade glioma. Nat. Commun. 2014, 5, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, N.S.; Chan, A.S.; Yee, J.D.; Yee, R.K. TRPM7 and TRPM8 ion channels in pancreatic adenocarcinoma: Potential roles as cancer biomarkers and targets. Scientifica 2012, 2012, 1–8. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Mutational Drivers | Calcium Alterations | Calcium Altering Therapies | References |
|---|---|---|---|---|
| Lung | EGFR | ↑Orai1 | Cisplatin | |
| KRAS | ↑TRPC1,3,4,6 | Doxorubicin | ||
| ALK | Afitinib | [22,74,87] | ||
| Breast | HER2 | ↑TRPV6 | Cisplatin | |
| BRCA1/2 | Tamoxifen | |||
| PTEN | TRPV6 Inhibitor | [83,85,86] | ||
| Colon | APC | ↑Stim1/Orai1 | Stim1/Orai1 Inhibitors | |
| KRAS | ↑SERCA2 | Thapsigargin | ||
| BRAF | ↑IP3 | 5-FU | [90,91,92] | |
| Prostate | FOXA1 | |||
| TP53 | ↑TRPV1 | Paclitaxel | ||
| PTEN | ↑TRPV2 | Cisplatin | [93,94] | |
| Pancreatic | KRAS | ↑TRPM7 | 5-FU | |
| TP53 | ↑Orai1 | [95,98] | ||
| Glioblastoma | LOH Loss | ↑TRPV1 | T-Type Calcium Channel Blocker Combined with TMZ | |
| ↑TRPV2 | [96,97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, C.A.; Hazlehurst, L.A. Role of Calcium Homeostasis in Modulating EMT in Cancer. Biomedicines 2021, 9, 1200. https://doi.org/10.3390/biomedicines9091200
Jones CA, Hazlehurst LA. Role of Calcium Homeostasis in Modulating EMT in Cancer. Biomedicines. 2021; 9(9):1200. https://doi.org/10.3390/biomedicines9091200
Chicago/Turabian StyleJones, Clark A., and Lori A. Hazlehurst. 2021. "Role of Calcium Homeostasis in Modulating EMT in Cancer" Biomedicines 9, no. 9: 1200. https://doi.org/10.3390/biomedicines9091200