
Autophagy, Mesenchymal Stem Cell Differentiation, and Secretion


Abstract
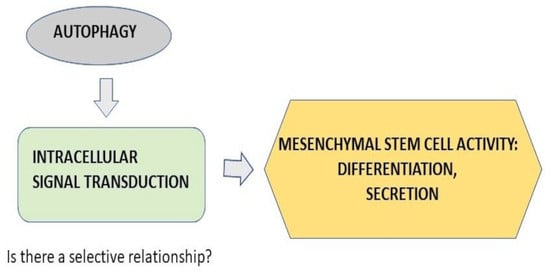
:
1. Introduction
2. Mesenchymal Stem Cells (MSC)
3. Autophagy: General Aspects
4. MSC Differentiation and Autophagy
5. Immunomodulatory Activity of MSC
6. Some Signal Processes Involving Autophagy
6.1. Notch
6.2. Hypoxia Inducible Factor (HIF-1α)
6.3. Nrf2/Keap1 Axis
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mazo, M.M.; Planat-Bénard, V.; Abizanda, G.; Pelacho, B.; Léobon, B.; Gavira, J.J.; Penuelas, I.; Cemborain, A.; Penicaud, L.; Laharrague, P.; et al. Transplantation of adipose derived stromal cells is associated with functional improvement in a rat model of chronic myocardial infarction. Eur. J. Heart Fail. 2008, 10, 454–462. [Google Scholar] [CrossRef] [Green Version]
- Wollert, K.C.; Drexler, H. Cell therapy for the treatment of coronary heart disease: A critical appraisal. Nat. Rev. Cardiol. 2010, 7, 204–215. [Google Scholar] [CrossRef]
- Guzzo, R.M.; Gibson, J.; Xu, R.-H.; Lee, F.Y.; Drissi, H. Efficient differentiation of human iPSC-derived mesenchymal stem cells to chondroprogenitor cells. J. Cell. Biochem. 2012, 114, 480–490. [Google Scholar] [CrossRef]
- Gao, W.-X.; Sun, Y.-Q.; Shi, J.; Li, C.-L.; Fang, S.-B.; Wang, D.; Deng, X.-Q.; Wen, W.; Fu, Q.-L. Effects of mesenchymal stem cells from human induced pluripotent stem cells on differentiation, maturation, and function of dendritic cells. Stem Cell Res. Ther. 2017, 8, 48. [Google Scholar] [CrossRef] [Green Version]
- Kusuma, G.D.; Carthew, J.R.; Lim, R.; Frith, J.E. Effect of the Microenvironment on Mesenchymal Stem Cell Paracrine Signaling: Opportunities to Engineer the Therapeutic Effect. Stem Cells Dev. 2017, 26, 617–631. [Google Scholar] [CrossRef]
- Meirelles, L.D.S.; Chagastelles, P.C.; Nardi, N.B. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci. 2006, 119, 2204–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, F.-J.; Tuan, R.S.; Cheung, K.M.; Leung, V.Y. Concise Review: The Surface Markers and Identity of Human Mesenchymal Stem Cells. Stem Cells 2014, 32, 1408–1419. [Google Scholar] [CrossRef]
- Lin, C.S.; Xin, Z.C.; Dai, J.; Lue, T.F. Commonly used mesenchymal stem cell markers and tracking labels: Limitations and challenges. Histol. Histopathol. 2013, 28, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, G.; Fox, J.; Ashton, B.; Middleton, J. Concise review: Mesenchymal stem cells: Their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells 2007, 25, 2739–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nombela-Arrieta, C.; Ritz, J.; Silberstein, L.E. The elusive nature and function of mesenchymal stem cells. Nat. Rev. Mol. Cell Biol. 2011, 12, 126–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.S.; Choi, Y.; Kim, H.-S.; Kim, H.O. Comparison of molecular profiles of human mesenchymal stem cells derived from bone marrow, umbilical cord blood, placenta and adipose tissue. Int. J. Mol. Med. 2015, 37, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, Y.; Gou, W.; Lu, Q.; Peng, J.; Lu, S. Role of mesenchymal stem cells in bone regeneration and fracture repair: A review. Int. Orthop. 2013, 37, 2491–2498. [Google Scholar] [CrossRef] [PubMed]
- Rebelatto, C.K.; Aguiar, A.M.; Moretão, M.P.; Senegaglia, A.C.; Hansen, P.; Barchiki, F.; Oliveira, J.M.; Martins, J.; Kuligovski, C.; Mansur, F.; et al. Dissimilar Differentiation of Mesenchymal Stem Cells from Bone Marrow, Umbilical Cord Blood, and Adipose Tissue. Exp. Biol. Med. 2008, 233, 901–913. [Google Scholar] [CrossRef]
- Alvarez-Dolado, M.; Pardal, R.; García-Verdugo, J.M.; Fike, J.R.; Lee, H.O.; Pfeffer, K.; Lois, C.; Morrison, S.; Alvarez-Buylla, A. Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature 2003, 425, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Meirelles, L.D.S.; Fontes, A.M.; Covas, D.T.; Caplan, A.I. Mechanisms involved in the therapeutic properties of mesenchymal stem cells. Cytokine Growth Factor Rev. 2009, 20, 419–427. [Google Scholar] [CrossRef]
- Hocking, A.M.; Gibran, N.S. Mesenchymal stem cells: Paracrine signaling and differentiation during cutaneous wound repair. Exp. Cell Res. 2010, 316, 2213–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Ami, E.; Berrih-Aknin, S.; Miller, A. Mesenchymal stem cells as an immunomodulatory therapeutic strategy for autoimmune diseases. Autoimmun. Rev. 2011, 10, 410–415. [Google Scholar] [CrossRef]
- Caplan, A.I.; Correa, D. The MSC: An Injury Drugstore. Cell Stem Cell 2011, 9, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Bieback, K.; Wuchter, P.; Besser, D.; Franke, W.; Becker, M.; Ott, M.; Pacher, M.; Ma, N.; Stamm, C.; Klüter, H.; et al. Mesenchymal stromal cells (MSCs): Science and f(r)iction. J. Mol. Med. 2012, 90, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Prockop, D.J.; Oh, J.Y. Medical therapies with adult stem/progenitor cells (MSCs): A backward journey from dramatic results in vivo to the cellular and molecular explanations. J. Cell. Biochem. 2011, 113, 1460–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronckaers, A.; Hilkens, P.; Martens, W.; Gervois, P.; Ratajczak, J.; Struys, T.; Lambrichts, I. Mesenchymal stem/stromal cells as a pharmacological and therapeutic approach to accelerate angiogenesis. Pharmacol. Ther. 2014, 143, 181–196. [Google Scholar] [CrossRef]
- Heo, J.S.; Choi, Y.; Kim, H.O. Adipose-Derived Mesenchymal Stem Cells Promote M2 Macrophage Phenotype through Exosomes. Stem Cells Int. 2019, 2019, 7921760. [Google Scholar] [CrossRef]
- Farmer, S.R. Transcriptional control of adipocyte formation. Cell Metab. 2006, 4, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkovskiy, A.; Stensløkken, K.-O.; Vaage, I.J. Osteoblast Differentiation at a Glance. Med. Sci. Monit. Basic Res. 2016, 22, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, A.W.; Marcon, B.H.; Dallagiovanna, B.; Shigunov, P. Adipogenesis, Osteogenesis, and Chondrogenesis of Human Mesenchymal Stem/Stromal Cells: A Comparative Transcriptome Approach. Front. Cell Dev. Biol. 2020, 8, 561. [Google Scholar] [CrossRef]
- Bassi, J.; Almeida, D.; Vieira, P.; Câmara, N.O.S. Exploring the Role of Soluble Factors Associated with Immune Regulatory Properties of Mesenchymal Stem Cells. Stem Cell Rev. Rep. 2011, 8, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Kachgal, S.; Putnam, A.J. Mesenchymal stem cells from adipose and bone marrow promote angiogenesis via distinct cytokine and protease expression mechanisms. Angiogenesis 2010, 14, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.K.; Chullikana, A.; Parakh, R.; Desai, S.; Das, A.; Gottipamula, S.; Krishnamurthy, S.; Anthony, N.; Pherwani, A.; Majumdar, A.S. A double blind randomized placebo controlled phase I/II study assessing the safety and efficacy of allogeneic bone marrow derived mesenchymal stem cell in critical limb ischemia. J. Transl. Med. 2013, 11, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, P.-O.; Schwarcz, E.; Korsgren, O.; Le Blanc, K. Preserved β-Cell Function in Type 1 Diabetes by Mesenchymal Stromal Cells. Diabetes 2014, 64, 587–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagnozzi, R.J.; Maillet, M.; Sargent, M.A.; Khalil, H.; Johansen, A.K.Z.; Schwanekamp, J.A.; York, A.J.; Huang, V.; Nahrendorf, M.; Sadayappan, S.; et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 2019, 577, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.K.; Kondziolka, D.; Wechsler, L.R.; Lunsford, L.D.; Coburn, M.L.; Billigen, J.B.; Kim, A.S.; Johnson, J.N.; Bates, D.; King, B.; et al. Clinical Outcomes of Transplanted Modified Bone Marrow–Derived Mesenchymal Stem Cells in Stroke: A Phase 1/2a Study. Stroke 2016, 47, 1817–1824. [Google Scholar] [CrossRef] [Green Version]
- Waterman, R.S.; Tomchuck, S.L.; Henkle, S.L.; Betancourt, A.M. A New Mesenchymal Stem Cell (MSC) Paradigm: Polarization into a Pro-Inflammatory MSC1 or an Immunosuppressive MSC2 Phenotype. PLoS ONE 2010, 5, e10088. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R.S.; Morgenweck, J.; Nossaman, B.D.; Scandurro, A.E.; Scandurro, S.A.; Betancourt, A.M. Anti-Inflammatory Mesenchymal Stem Cells (MSC2) Attenuate Symptoms of Painful Diabetic Peripheral Neuropathy. Stem Cells Transl. Med. 2012, 1, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Dergilev, K.V.; Shevchenko, E.K.; Tsokolaeva, Z.I.; Beloglazova, I.B.; Zubkova, E.S.; Boldyreva, M.A.; Menshikov, M.Y.; Ratner, E.I.; Penkov, D.; Parfyonova, Y.V. Cell Sheet Comprised of Mesenchymal Stromal Cells Overexpressing Stem Cell Factor Promotes Epicardium Activation and Heart Function Improvement in a Rat Model of Myocardium Infarction. Int. J. Mol. Sci. 2020, 21, 9603. [Google Scholar] [CrossRef] [PubMed]
- Boldyreva, M.A.; Shevchenko, E.K.; Molokotina, Y.D.; Makarevich, P.I.; Beloglazova, I.B.; Zubkova, E.S.; Dergilev, K.V.; Tsokolaeva, Z.I.; Penkov, D.; Hsu, M.-N.; et al. Transplantation of Adipose Stromal Cell Sheet Producing Hepatocyte Growth Factor Induces Pleiotropic Effect in Ischemic Skeletal Muscle. Int. J. Mol. Sci. 2019, 20, 3088. [Google Scholar] [CrossRef] [Green Version]
- Heldring, N.; Mäger, I.; Wood, M.J.; Le Blanc, K.; Andaloussi, S.E. Therapeutic Potential of Multipotent Mesenchymal Stromal Cells and Their Extracellular Vesicles. Hum. Gene Ther. 2015, 26, 506–517. [Google Scholar] [CrossRef]
- Zubkova, E.; Beloglazova, I.B.; Evtushenko, E.; Kopylov, A.T.; Shevchenko, E.K.; Dergilev, K.V.; Ratner, E.I.; Parfenova, E.V.; Men’Shikov, M.Y. Application of Adeno-Associated Virus Vectors for Engineering SCF-Containing Extracellular Vesicles of Mesenchymal Stromal Cells. Bull. Exp. Biol. Med. 2019, 166, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Ahfeldt, T.; Schinzel, R.T.; Lee, Y.-K.; Hendrickson, D.; Kaplan, A.; Lum, D.H.; Camahort, R.; Xia, F.; Shay, J.; Rhee, E.P.; et al. Programming human pluripotent stem cells into white and brown adipocytes. Nat. Cell Biol. 2012, 14, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Li, K.-C.; Hu, Y.-C. Cartilage Tissue Engineering: Recent Advances and Perspectives from Gene Regulation/Therapy. Adv. Heal. Mater. 2015, 4, 948–968. [Google Scholar] [CrossRef]
- Yeh, T.-S.; Fang, Y.-H.D.; Lu, C.-H.; Chiu, S.-C.; Yeh, C.-L.; Yen, T.-C.; Parfyonova, Y.; Hu, Y.-C. Baculovirus-transduced, VEGF-expressing adipose-derived stem cell sheet for the treatment of myocardium infarction. Biomaterials 2014, 35, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and Autophagy-Related Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.-W.; Li, J.; Bao, J.-K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2011, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2013, 24, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, C.; Yoshimori, T.; Tooze, S. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y. Regulation and Function of Uncoordinated-51 Like Kinase Proteins. Antioxid. Redox Signal. 2012, 17, 775–785. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef]
- Kuma, A.; Mizushima, N.; Ishihara, N.; Ohsumi, Y. Formation of the ∼350-kDa Apg12-Apg5·Apg16 Multimeric Complex, Mediated by Apg16 Oligomerization, Is Essential for Autophagy in Yeast. J. Biol. Chem. 2002, 277, 18619–18625. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J.; et al. Deacetylation of Nuclear LC3 Drives Autophagy Initiation under Starvation. Mol. Cell 2015, 57, 456–466. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippai, M.; Lőw, P. The Role of the Selective Adaptor p62 and Ubiquitin-Like Proteins in Autophagy. BioMed Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.I.; Gill, D.J.; Perisic, O.; Quinn, M.; Williams, R.L. PB1 Domain-Mediated Heterodimerization in NADPH Oxidase and Signaling Complexes of Atypical Protein Kinase C with Par6 and p62. Mol. Cell 2003, 12, 39–50. [Google Scholar] [CrossRef]
- Nakamura, K.; Kimple, A.J.; Siderovski, D.; Johnson, G.L. PB1 Domain Interaction of p62/Sequestosome 1 and MEKK3 Regulates NF-κB Activation. J. Biol. Chem. 2010, 285, 2077–2089. [Google Scholar] [CrossRef] [Green Version]
- Axe, E.L.; Walker, S.; Manifava, M.; Chandra, P.; Roderick, H.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria Supply Membranes for Autophagosome Biogenesis during Starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.-I.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 Association with the ULK1–Atg13–FIP200 Complex Required for Autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Shao, S.H.; Xu, Z.-X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1–AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Weissman, I.L. Stem cells are units of natural selection for tissue formation, for germline development, and in cancer development. Proc. Natl. Acad. Sci. USA 2015, 112, 8922–8928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPARγ Is Required for the Differentiation of Adipose Tissue In Vivo and In Vitro. Mol. Cell 1999, 4, 611–617. [Google Scholar] [CrossRef]
- Komori, T. Regulation of Osteoblast Differentiation by Runx2. Adv. Exp. Med. Biol. 2010, 658, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 2009, 119, 3329–3339. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.S.; Feinberg, M.W.; Watanabe, M.; Gray, S.; Haspel, R.L.; Denkinger, D.J.; Kawahara, R.; Hauner, H.; Jain, M.K. The Krüppel-like Factor KLF2 Inhibits Peroxisome Proliferator-activated Receptor-γ Expression and Adipogenesis. J. Biol. Chem. 2003, 278, 2581–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Huang, J.-X.; Liu, Y.; Li, X.; Zhou, S.-R.; Qian, S.-W.; Liu, Y.; Zhu, H.; Huang, H.-Y.; Dang, Y.-J.; et al. Transactivation of Atg4b by C/EBPβ Promotes Autophagy To Facilitate Adipogenesis. Mol. Cell. Biol. 2013, 33, 3180–3190. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Ha, J.-H.; Okla, M.; Chung, S. Activation of autophagy and AMPK by gamma-tocotrienol suppresses the adipogenesis in human adipose derived stem cells. Mol. Nutr. Food Res. 2013, 58, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petherick, K.J.; Williams, A.; Lane, J.; Ordóñez-Morán, P.; Huelsken, J.; Collard, T.J.; Smartt, H.J.; Batson, J.; Malik, K.; Paraskeva, C.; et al. Autolysosomal β-catenin degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J. 2013, 32, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Cao, W.; Bao, L.; Zuo, W.; Xie, G.; Cai, T.; Fu, W.; Zhang, J.; Wu, W.; Zhang, X.; et al. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat. Cell Biol. 2010, 12, 781–790. [Google Scholar] [CrossRef]
- Prestwich, T.C.; MacDougald, O.A. Wnt/β-catenin signaling in adipogenesis and metabolism. Curr. Opin. Cell Biol. 2007, 19, 612–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, L.J.; Bonnycastle, L.L.; Willer, C.J.; Sprau, A.G.; Jackson, A.U.; Narisu, N.; Duren, W.L.; Chines, P.S.; Stringham, H.M.; Erdos, M.R.; et al. Association of Transcription Factor 7-Like 2 (TCF7L2) Variants With Type 2 Diabetes in a Finnish Sample. Diabetes 2006, 55, 2649–2653. [Google Scholar] [CrossRef] [Green Version]
- Reilly, S.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-S.; Lu, C.-C.; Kuo, S.-C.; Hsu, Y.-M.; Tsai, S.-C.; Chen, S.-Y.; Chen, Y.-T.; Lin, Y.-J.; Huang, Y.-C.; Chen, C.-J.; et al. Autophagy and its link to type II diabetes mellitus. BioMedicine 2017, 7, 8. [Google Scholar] [CrossRef] [Green Version]
- Kovsan, J.; Blüher, M.; Tarnovscki, T.; Klöting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schön, M.; et al. Altered Autophagy in Human Adipose Tissues in Obesity. J. Clin. Endocrinol. Metab. 2011, 96, E268–E277. [Google Scholar] [CrossRef]
- Vidoni, C.; Ferraresi, A.; Secomandi, E.; Vallino, L.; Gardin, C.; Zavan, B.; Mortellaro, C.; Isidoro, C. Autophagy drives osteogenic differentiation of human gingival mesenchymal stem cells. Cell Commun. Signal. 2019, 17, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Zhuo, N.; Li, Y.; Zhao, W.; Jiang, D. Autophagy promotes osteogenic differentiation of human bone marrow mesenchymal stem cell derived from osteoporotic vertebrae. Biochem. Biophys. Res. Commun. 2017, 488, 46–52. [Google Scholar] [CrossRef]
- Ma, Y.; Qi, M.; An, Y.; Zhang, L.; Yang, R.; Doro, D.H.; Liu, W.; Jin, Y. Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging Cell 2017, 17, e12709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Bennett, C.N.; Gerin, I.; Rapp, L.A.; Hankenson, K.D.; MacDougald, O.A. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2007, 282, 14515–14524. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.T.; Reuveny, S.; Oh, S.K.-W. Human mesenchymal stem cell therapy for cartilage repair: Review on isolation, expansion, and constructs. Stem Cell Res. 2020, 44, 101738. [Google Scholar] [CrossRef] [PubMed]
- Horigome, Y.; Ida-Yonemochi, H.; Waguri, S.; Shibata, S.; Endo, N.; Komatsu, M. Loss of autophagy in chondrocytes causes severe growth retardation. Autophagy 2019, 16, 501–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Luo, H.; Wang, R.; Kang, Y.; Liao, W.; Sun, Y.; Chen, G.; Shao, L. Rapamycin-Induced Autophagy Promotes the Chondrogenic Differentiation of Synovium-Derived Mesenchymal Stem Cells in the Temporomandibular Joint in Response to IL-1β. BioMed Res. Int. 2020, 2020, 4035306. [Google Scholar] [CrossRef] [PubMed]
- Kirton, J.P.; Crofts, N.J.; George, S.J.; Brennan, K.; Canfield, A.E. Wnt/β-Catenin Signaling Stimulates Chondrogenic and Inhibits Adipogenic Differentiation of Pericytes: Potential Relevance to Vascular Disease? Circ. Res. 2007, 101, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.; Dahlke, M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of Action of Living, Apoptotic, and Dead MSCs. Front. Immunol. 2019, 10, 1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurora, A.B.; Porrello, E.; Tan, W.; Mahmoud, A.I.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; Olson, E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 2014, 124, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Saldaña, L.; Bensiamar, F.; Vallés, G.; Mancebo, F.J.; García-Rey, E.; Vilaboa, N. Immunoregulatory potential of mesenchymal stem cells following activation by macrophage-derived soluble factors. Stem Cell Res. Ther. 2019, 10, 58. [Google Scholar] [CrossRef]
- Chung, E.; Son, Y. Crosstalk between mesenchymal stem cells and macrophages in tissue repair. Tissue Eng. Regen. Med. 2014, 11, 431–438. [Google Scholar] [CrossRef]
- Wang, B.; Lin, Y.; Hu, Y.; Shan, W.; Liu, S.; Xu, Y.; Zhang, H.; Cai, S.; Yu, X.; Cai, Z.; et al. mTOR inhibition improves the immunomodulatory properties of human bone marrow mesenchymal stem cells by inducing COX-2 and PGE2. Stem Cell Res. Ther. 2017, 8, 292. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Tan, W.; Ji, J.; Feng, G.; Meng, Y.; Da, Z.; Guo, G.; Xia, Y.; Zhu, X.; Shi, G.; et al. Rapamycin reverses the senescent phenotype and improves immunoregulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling path-way. Aging 2016, 8, 1102–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Cen, S.; Wang, P.; Xie, Z.; Liu, Z.; Deng, W.; Su, H.; Wu, X.; Wang, S.; Li, J.; et al. Autophagy Improves the Immunosuppression of CD4+ T Cells by Mesenchymal Stem Cells Through Transforming Growth Factor-β1. Stem Cells Transl. Med. 2016, 5, 1496–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cen, S.; Wang, P.; Xie, Z.; Yang, R.; Li, J.; Liu, Z.; Wang, S.; Wu, X.; Liu, W.; Li, M.; et al. Autophagy enhances mesenchymal stem cell-mediated CD4+ T cell migration and differentiation through CXCL8 and TGF-β1. Stem Cell Res. Ther. 2019, 10, 265. [Google Scholar] [CrossRef] [Green Version]
- Hori, K.; Sen, A.; Artavanis-Tsakonas, S. Notch signaling at a glance. J. Cell Sci. 2013, 126, 2135–2140. [Google Scholar] [CrossRef] [Green Version]
- Song, B.-Q.; Chi, Y.; Li, X.; Du, W.; Han, Z.-B.; Tian, J.-J.; Li, J.-J.; Chen, F.; Wu, H.; Han, L.-X.; et al. Inhibition of Notch Signaling Promotes the Adipogenic Differentiation of Mesenchymal Stem Cells Through Autophagy Activation and PTEN-PI3K/AKT/mTOR Pathway. Cell. Physiol. Biochem. 2015, 36, 1991–2002. [Google Scholar] [CrossRef]
- Semenova, D.; Bogdanova, M.; Kostina, A.; Golovkin, A.; Kostareva, A.; Malashicheva, A. Dose-dependent mechanism of Notch action in promoting osteogenic differentiation of mesenchymal stem cells. Cell Tissue Res. 2019, 379, 169–179. [Google Scholar] [CrossRef]
- Mitterberger, M.C.; Lechner, S.; Mattesich, M.; Kaiser, A.; Probst, D.; Wenger, N.; Pierer, G.; Zwerschke, W. DLK1(PREF1) is a negative regulator of adipogenesis in CD105+/CD90+/CD34+/CD31−/FABP4− adipose-derived stromal cells from subcu-taneous abdominal fat pats of adult women. Stem Cell Res. 2012, 9, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Sul, H.S. Minireview: Pref-1: Role in Adipogenesis and Mesenchymal Cell Fate. Mol. Endocrinol. 2009, 23, 1717–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, B.; Li, F.; Han, J.; Fang, J.; Xu, L.; Sun, C.; Hua, T.; Zhang, Z.; Feng, Z.; Jiang, X. Hif-1α Overexpression Improves Transplanted Bone Mesenchymal Stem Cells Survival in Rat MCAO Stroke Model. Front. Mol. Neurosci. 2017, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Li, X.; Tan, R.; An, J.; Cai, Z.; Hu, X.; Wang, F.; Wang, H.; Lu, C.; Lu, H. HIF-1α/Beclin1-Mediated Autophagy Is Involved in Neuroprotection Induced by Hypoxic Preconditioning. J. Mol. Neurosci. 2018, 66, 238–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhang, Z.; Xiong, W.; Zhang, L.; Xiong, Y.; Li, N.; He, H.; Du, Y.; Liu, Y. Hypoxia-inducible factor-1α promotes endometrial stromal cells migration and invasion by upregulating autophagy in endometriosis. Reproduction 2017, 153, 809–820. [Google Scholar] [CrossRef]
- Wagegg, M.; Gaber, T.; Lohanatha, F.L.; Hahne, M.; Strehl, C.; Fangradt, M.; Tran, C.L.; Schönbeck, K.; Hoff, P.; Ode, A.; et al. Hypoxia Promotes Osteogenesis but Suppresses Adipogenesis of Human Mesenchymal Stromal Cells in a Hypoxia-Inducible Factor-1 Dependent Manner. PLoS ONE 2012, 7, e46483. [Google Scholar] [CrossRef] [Green Version]
- Belibi, F.; Zafar, I.; Ravichandran, K.; Segvic, A.B.; Jani, A.; Ljubanovic, D.G.; Edelstein, C.L. Hypoxia-inducible factor-1α (HIF-1α) and autophagy in polycystic kidney disease (PKD). Am. J. Physiol.-Ren. Physiol. 2011, 300, F1235–F1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Mutter, F.E.; Park, B.K.; Copple, I.M. Value of monitoring Nrf2 activity for the detection of chemical and oxidative stress. Biochem. Soc. Trans. 2015, 43, 657–662. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Davies, K.J.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free. Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keum, Y.-S.; Choi, B.Y. Molecular and Chemical Regulation of the Keap1-Nrf2 Signaling Pathway. Molecules 2014, 19, 10074–10089. [Google Scholar] [CrossRef] [Green Version]
- Ornatowski, W.; Lu, Q.; Yegambaram, M.; Garcia, A.E.; Zemskov, E.A.; Maltepe, E.; Fineman, J.R.; Wang, T.; Black, S.M. Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 2020, 36, 101679. [Google Scholar] [CrossRef] [PubMed]
- Szumiel, I. Autophagy, reactive oxygen species and the fate of mammalian cells. Free. Radic. Res. 2010, 45, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.; Johansen, T. p62/SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-driven Gene Transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Chen, Z.; She, C.; Lin, Y.; Hong, Y.; Shi, L.; Zhang, Y.; Cao, P.; Xu, X. Four-octyl itaconate activates Nrf2 cascade to protect osteoblasts from hydrogen peroxide-induced oxidative injury. Cell Death Dis. 2020, 11, 772. [Google Scholar] [CrossRef] [PubMed]
- Marchese, P.; Mahajan, N.; O’Connell, E.; Fearnhead, H.; Tuohy, M.; Krawczyk, J.; Thomas, O.P.; Barry, F.; Murphy, M.J. A Novel High-Throughput Screening Platform Identifies Itaconate Derivatives from Marine Penicillium antarcticum as Inhibitors of Mesenchymal Stem Cell Differentiation. Mar. Drugs 2020, 18, 192. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Wang, H.; Zhai, Y.; Park, H.; Wang, J.; Ji, F.; Zhang, Z. Downregulation of Nrf2 promotes autophagy-dependent osteoblastic differentiation of adipose-derived mesenchymal stem cells. Exp. Cell Res. 2016, 349, 221–229. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| MSC Activity | Markers | Responsible Mechanisms | Modulators | Reference |
|---|---|---|---|---|
| MSC differentiation | ||||
| Adipogenic | Glut4, Perilipin-2, PGC1α, Pref1, UCP-1, aP2 | CEBPα, PPARγ | Klf2/Klf3 Pref1 | [25] |
| Osteogenic | ALPP, SPARC, collagen I | RUNX2, Osterix | LY3023414 | [26] |
| Chondrogenic | Annexin A6, CD44, CD151, ITM2A, collagen II/IV | FAM20B, FoxC1, Fox C2/SOX9 | SOX9, Il-1β | [27] |
| MSC secreted factors | ||||
| Tissue repair/angiogenesis | VEGF, HGF, EGF, TNFα, MIP-1, TIMPs, IL6, IL8 | Pro-/anti-inflammatory signaling, MAPK kinases | Cell signaling inhibitors | [23,25] |
| Immunomodulation | IDO, TGFβ, HGF, PGE2 | Pro-/anti-inflammatory signaling, MAPK kinases | Cell signaling inhibitors | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menshikov, M.; Zubkova, E.; Stafeev, I.; Parfyonova, Y. Autophagy, Mesenchymal Stem Cell Differentiation, and Secretion. Biomedicines 2021, 9, 1178. https://doi.org/10.3390/biomedicines9091178
Menshikov M, Zubkova E, Stafeev I, Parfyonova Y. Autophagy, Mesenchymal Stem Cell Differentiation, and Secretion. Biomedicines. 2021; 9(9):1178. https://doi.org/10.3390/biomedicines9091178
Chicago/Turabian StyleMenshikov, Mikhail, Ekaterina Zubkova, Iuri Stafeev, and Yelena Parfyonova. 2021. "Autophagy, Mesenchymal Stem Cell Differentiation, and Secretion" Biomedicines 9, no. 9: 1178. https://doi.org/10.3390/biomedicines9091178