
Targeted Inhibition of Anti-Inflammatory Regulator Nrf2 Results in Breast Cancer Retardation In Vitro and In Vivo

 ,
,  ,
,
Abstract
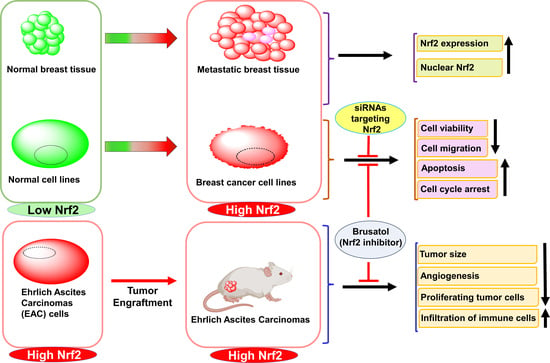
:
1. Introduction
2. Materials and Methods
2.1. Collection of Tumor Samples and Used Chemicals
2.2. Collection of Breast Cancer Tissues, Construction of a Tissue Array, and Immunohistochemistry (IHC)
2.3. Isolation, Estimation of Total Protein, and Western Blotting Analysis
2.4. Measurement of NQO1 Activity
2.5. siRNA Mediated Nrf2 Knockdown Using Lipofectamine RNAi Max Reagent
2.6. Analysis of Gene Expression Using qRT-PCR and Gel Electrophoresis of Total RNA
2.7. Cytotoxic Potential of Nrf2 Inhibitor Brusatol
2.8. Cell Cycle Analysis Using Propidium Iodide (PI) Staining
2.9. Assessment of Cell Migration Using Scratch Assay
2.10. Apoptosis Assay with Acridine Orange (AO) and Ethidium Bromide (EtBr)
2.11. Assessment of Lymphocytic Infiltration in Solid Tumors
2.12. In-Vivo Evaluation of Brusatol Effects in Swiss Albino Mouse Model
2.13. Statistical Analysis
3. Results
3.1. Diversity of Nrf2 Expression Pattern in BC Tumors and Cell Lines
3.2. siRNA-Dependent Inhibition of Nrf2 Reduced BC Cells Viability and Sensitized Cells to Cisplatin In Vitro
3.3. Pharmacological Inhibition of Nrf2 Using Brusatol Blocked BC Cell Growth and Migration In Vitro
3.4. Genetic and Pharmacological Inhibition of Nrf2 Induced Accumulation of Cells in Sub-G0-G1 and G2/M Phases of Cell Cycle
3.5. Nrf2 Inhibitor Brusatol Stimulates BC Cell Death
3.6. Intraperitoneal Administration of Brusatol Inhibited EAC Solid Tumors Development in Mice In Vivo
3.7. Brusatol Treatment Reduced Nrf2 Activity/Expression and the Activity of Nrf2-Target Gene NQO1 in Tumors In Vivo
3.8. Brusatol-Induced Tumor Growth Inhibition Is Mediated by Decreased Expression of Ki67, CD31, and Enhanced Lymphocyte Invasion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, S.; Batoo, S.; Okuno, S.; Gluck, S. Immunotherapy in breast cancer. J. Carcinog. 2019, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Masoud, V.; Pages, G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J. Clin. Oncol. 2017, 8, 120–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, B.; Fuchs, B.C.; Medarova, Z. New Directions in the Study and Treatment of Metastatic Cancer. Front. Oncol. 2018, 8, 258. [Google Scholar] [CrossRef] [Green Version]
- Koury, J.; Lucero, M.; Cato, C.; Chang, L.; Geiger, J.; Henry, D.; Hernandez, J.; Hung, F.; Kaur, P.; Teskey, G.; et al. Immunotherapies: Exploiting the Immune System for Cancer Treatment. J. Immunol. Res. 2018, 2018, 9585614. [Google Scholar] [CrossRef]
- Cai, F.; Luis, M.A.F.; Lin, X.; Wang, M.; Cai, L.; Cen, C.; Biskup, E. Anthracycline-induced cardiotoxicity in the chemotherapy treatment of breast cancer: Preventive strategies and treatment. Mol. Clin. Oncol. 2019, 11, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Saloustros, E.; Mavroudis, D.; Georgoulias, V. Paclitaxel and docetaxel in the treatment of breast cancer. Expert Opin. Pharmacother. 2008, 9, 2603–2616. [Google Scholar] [CrossRef]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [CrossRef]
- Hernandez-Aya, L.F.; Ma, C.X. Chemotherapy principles of managing stage IV breast cancer in the United States. Chin. Clin. Oncol. 2016, 5, 42. [Google Scholar] [CrossRef]
- Tong, C.W.S.; Wu, M.; Cho, W.C.S.; To, K.K.W. Recent Advances in the Treatment of Breast Cancer. Front. Oncol. 2018, 8, 227. [Google Scholar] [CrossRef] [Green Version]
- Taherkhani, M.; Mahjoub, S.; Moslemi, D.; Karkhah, A. Three cycles of AC chemotherapy regimen increased oxidative stress in breast cancer patients: A clinical hint. Caspian J. Intern. Med. 2017, 8, 264–268. [Google Scholar] [CrossRef]
- Cocconi, G.; Bisagni, G.; Bella, M.; Acito, L.; Anastasi, P.; Carpi, A.; Di Costanzo, F.; Frassoldati, A.; Mosconi, A.; Borrini, A.; et al. Comparison of CMF (cyclophosphamide, methotrexate, and 5-fluorouracil) with a rotational crossing and a sequential intensification regimen in advanced breast cancer: A prospective randomized study. Am. J. Clin. Oncol. 1999, 22, 593–600. [Google Scholar] [CrossRef]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Fayanju, O.M.; Park, K.U.; Lucci, A. Molecular Genomic Testing for Breast Cancer: Utility for Surgeons. Ann. Surg. Oncol. 2018, 25, 512–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, N.; Al-Allak, A.; Fowler, C. Benefits of introduction of Oncotype DX((R)) testing. Ann. R Coll. Surg. Engl. 2019, 101, 55–59. [Google Scholar] [CrossRef]
- Wuerstlein, R.; Kates, R.; Gluz, O.; Grischke, E.M.; Schem, C.; Thill, M.; Hasmueller, S.; Kohler, A.; Otremba, B.; Griesinger, F.; et al. Strong impact of MammaPrint and BluePrint on treatment decisions in luminal early breast cancer: Results of the WSG-PRIMe study. Breast Cancer Res. Treat. 2019, 175, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef] [PubMed]
- Jessen, C.; Kress, J.K.C.; Baluapuri, A.; Hufnagel, A.; Schmitz, W.; Kneitz, S.; Roth, S.; Marquardt, A.; Appenzeller, S.; Ade, C.P.; et al. The transcription factor NRF2 enhances melanoma malignancy by blocking differentiation and inducing COX2 expression. Oncogene 2020, 39, 6841–6855. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Wang, H.J.; Bao, Q.C.; Wang, L.; Guo, T.K.; Chen, W.L.; Xu, L.L.; Zhou, H.S.; Bian, J.L.; Yang, Y.R.; et al. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 2016, 7, 73593–73606. [Google Scholar] [CrossRef] [Green Version]
- De Blasio, A.; Di Fiore, R.; Pratelli, G.; Drago-Ferrante, R.; Saliba, C.; Baldacchino, S.; Grech, G.; Scerri, C.; Vento, R.; Tesoriere, G. A loop involving NRF2, miR-29b-1-5p and AKT, regulates cell fate of MDA-MB-231 triple-negative breast cancer cells. J. Cell Physiol. 2020, 235, 629–637. [Google Scholar] [CrossRef]
- Qin, S.; He, X.; Lin, H.; Schulte, B.A.; Zhao, M.; Tew, K.D.; Wang, G.Y. Nrf2 inhibition sensitizes breast cancer stem cells to ionizing radiation via suppressing DNA repair. Free Radic. Biol. Med. 2021, 169, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Baniasadi, P.S.; Harris, I.S.; Silvester, J.; Inoue, S.; Snow, B.; Joshi, P.A.; Wakeham, A.; Molyneux, S.D.; Martin, B.; et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013, 210, 1529–1544. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, I.G.; Choi, B.H.; Ku, S.K.; Kwak, M.K. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: Implications for cancer stem cell resistance. Redox. Biol. 2018, 17, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Yu, S.; Chen, J.D.; Kong, A.N. The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains. Oncogene 2013, 32, 514–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-kappaB and Their Concerted Modulation in Cancer Pathogenesis and Progression. Cancers 2010, 2, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panieri, E.; Saso, L. Potential Applications of NRF2 Inhibitors in Cancer Therapy. Oxid. Med. Cell Longev. 2019, 2019, 8592348. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.P.; Winiarski, B.K.; Sutton, P.A.; Jones, R.P.; Ressel, L.; Duckworth, C.A.; Pritchard, D.M.; Lin, Z.X.; Fretwell, V.L.; Tweedle, E.M.; et al. The Nrf2 inhibitor brusatol is a potent antitumour agent in an orthotopic mouse model of colorectal cancer. Oncotarget 2018, 9, 27104–27116. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, P.; Csonka, T.; Kovacs, T.; Sari, Z.; Ujlaki, G.; Sipos, A.; Karanyi, Z.; Szeocs, D.; Hegedus, C.; Uray, K.; et al. Lithocholic Acid, a Metabolite of the Microbiome, Increases Oxidative Stress in Breast Cancer. Cancers 2019, 11, 1255. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Alcivar, A.L.; Ma, J.; Foo, T.K.; Zywea, S.; Mahdi, A.; Huo, Y.; Kensler, T.W.; Gatza, M.L.; Xia, B. NRF2 Induction Supporting Breast Cancer Cell Survival Is Enabled by Oxidative Stress-Induced DPP3-KEAP1 Interaction. Cancer Res. 2017, 77, 2881–2892. [Google Scholar] [CrossRef] [Green Version]
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Dou, Y.; Lin, G.; Li, Q.; Nie, J.; Chen, B.; Xie, J.; Su, Z.; Zeng, H.; Chen, J.; et al. The anti-hepatocellular carcinoma effect of Brucea javanica oil in ascitic tumor-bearing mice: The detection of brusatol and its role. Biomed. Pharmacother. 2021, 134, 111122. [Google Scholar] [CrossRef]
- Cai, S.J.; Liu, Y.; Han, S.; Yang, C. Brusatol, an NRF2 inhibitor for future cancer therapeutic. Cell Biosci. 2019, 9, 45. [Google Scholar] [CrossRef] [Green Version]
- Panieri, E.; Buha, A.; Telkoparan-Akillilar, P.; Cevik, D.; Kouretas, D.; Veskoukis, A.; Skaperda, Z.; Tsatsakis, A.; Wallace, D.; Suzen, S.; et al. Potential Applications of NRF2 Modulators in Cancer Therapy. Antioxidants 2020, 9, 193. [Google Scholar] [CrossRef] [Green Version]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preul, M.C.; Stratford, J.; Bertrand, G.; Feindel, W. Neurosurgeon as innovator: William V. Cone (1897–1959). J. Neurosurg. 1993, 79, 619–631. [Google Scholar] [CrossRef]
- Prashanth, T.; Avin, B.R.V.; Thirusangu, P.; Ranganatha, V.L.; Prabhakar, B.T.; Sharath Chandra, J.N.N.; Khanum, S.A. Synthesis of coumarin analogs appended with quinoline and thiazole moiety and their apoptogenic role against murine ascitic carcinoma. Biomed. Pharmacother. 2019, 112, 108707. [Google Scholar] [CrossRef] [PubMed]
- Goud, K.I.; Dayakar, S.; Vijayalaxmi, K.; Babu, S.J.; Reddy, P.V. Evaluation of HER-2/neu status in breast cancer specimens using immunohistochemistry (IHC) & fluorescence in-situ hybridization (FISH) assay. Indian J. Med. Res. 2012, 135, 312–317. [Google Scholar] [PubMed]
- Krishnamurthy, J.; Kumar, P.S. Significance of prognostic indicators in infiltrating duct carcinoma breast: Scenario in developing country. Indian J. Cancer 2016, 53, 34–38. [Google Scholar] [CrossRef]
- Gowda, R.; Madhunapantula, S.V.; Kuzu, O.F.; Sharma, A.; Robertson, G.P. Targeting multiple key signaling pathways in melanoma using leelamine. Mol. Cancer Ther. 2014, 13, 1679–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhunapantula, S.V.; Sharma, A.; Robertson, G.P. PRAS40 deregulates apoptosis in malignant melanoma. Cancer Res. 2007, 67, 3626–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochaska, H.J.; Santamaria, A.B. Direct measurement of NAD(P)H:quinone reductase from cells cultured in microtiter wells: A screening assay for anticarcinogenic enzyme inducers. Anal. Biochem. 1988, 169, 328–336. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: Twenty-something years on. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Aranda, P.S.; LaJoie, D.M.; Jorcyk, C.L. Bleach gel: A simple agarose gel for analyzing RNA quality. Electrophoresis 2012, 33, 366–369. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Gao, J.; Zheng, Y.; Wang, X.; Chen, C.; Cao, K.; Xu, J.; Li, Y.; Lu, W.; Liu, J.; et al. Zeaxanthin induces Nrf2-mediated phase II enzymes in protection of cell death. Cell Death Dis. 2014, 5, e1218. [Google Scholar] [CrossRef] [Green Version]
- Seng, S.; Avraham, H.K.; Birrane, G.; Jiang, S.; Li, H.; Katz, G.; Bass, C.E.; Zagozdzon, R.; Avraham, S. NRP/B mutations impair Nrf2-dependent NQO1 induction in human primary brain tumors. Oncogene 2009, 28, 378–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; An, J.; Ji, F.; Jiao, H.; Sun, H.; Zhou, D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem. Biophys. Res. Commun. 2008, 373, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Lister, A.; Nedjadi, T.; Kitteringham, N.R.; Campbell, F.; Costello, E.; Lloyd, B.; Copple, I.M.; Williams, S.; Owen, A.; Neoptolemos, J.P.; et al. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol. Cancer 2011, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef]
- Nam, S.T.; Hwang, J.H.; Kim, D.H.; Park, M.J.; Lee, I.H.; Nam, H.J.; Kang, J.K.; Kim, S.K.; Hwang, J.S.; Chung, H.K.; et al. Role of NADH: Quinone oxidoreductase-1 in the tight junctions of colonic epithelial cells. BMB Rep. 2014, 47, 494–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrickx, A.; Pierrot, N.; Tasiaux, B.; Schakman, O.; Kienlen-Campard, P.; De Smet, C.; Octave, J.N. Epigenetic regulations of immediate early genes expression involved in memory formation by the amyloid precursor protein of Alzheimer disease. PLoS ONE 2014, 9, e99467. [Google Scholar] [CrossRef]
- Madhunapantula, S.V.; Desai, D.; Sharma, A.; Huh, S.J.; Amin, S.; Robertson, G.P. PBISe, a novel selenium-containing drug for the treatment of malignant melanoma. Mol. Cancer Ther. 2008, 7, 1297–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana, E.A.; Kasinski, A.L. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio Protoc. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meerloo, J.; Kaspers, G.J.; Cloos, J. Cell sensitivity assays: The MTT assay. Methods Mol. Biol 2011, 731, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Hrgovic, I.; Doll, M.; Kleemann, J.; Wang, X.F.; Zoeller, N.; Pinter, A.; Kippenberger, S.; Kaufmann, R.; Meissner, M. The histone deacetylase inhibitor trichostatin a decreases lymphangiogenesis by inducing apoptosis and cell cycle arrest via p21-dependent pathways. BMC Cancer 2016, 16, 763. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Varankar, S.S.; Bapat, S.A. Migratory Metrics of Wound Healing: A Quantification Approach for in vitro Scratch Assays. Front. Oncol. 2018, 8, 633. [Google Scholar] [CrossRef]
- Chen, K.; Cheng, L.; Qian, W.; Jiang, Z.; Sun, L.; Zhao, Y.; Zhou, Y.; Zhao, L.; Wang, P.; Duan, W.; et al. Itraconazole inhibits invasion and migration of pancreatic cancer cells by suppressing TGF-beta/SMAD2/3 signaling. Oncol. Rep. 2018, 39, 1573–1582. [Google Scholar] [CrossRef] [Green Version]
- Jonkman, J.E.; Cathcart, J.A.; Xu, F.; Bartolini, M.E.; Amon, J.E.; Stevens, K.M.; Colarusso, P. An introduction to the wound healing assay using live-cell microscopy. Cell Adh. Migr. 2014, 8, 440–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grada, A.; Otero-Vinas, M.; Prieto-Castrillo, F.; Obagi, Z.; Falanga, V. Research Techniques Made Simple: Analysis of Collective Cell Migration Using the Wound Healing Assay. J. Investig. Dermatol. 2017, 137, e11–e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujani, M.; Jain, H.; Chauhan, V.; Agarwal, C.; Singh, K.; Singh, M. Evaluation of Tumor infiltrating lymphocytes in breast carcinoma and their correlation with molecular subtypes, tumor grade and stage. Breast Dis. 2020, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Thirusangu, P.; Vigneshwaran, V.; Prashanth, T.; Vijay Avin, B.R.; Malojirao, V.H.; Rakesh, H.; Khanum, S.A.; Mahmood, R.; Prabhakar, B.T. BP-1T, an antiangiogenic benzophenone-thiazole pharmacophore, counteracts HIF-1 signalling through p53/MDM2-mediated HIF-1alpha proteasomal degradation. Angiogenesis 2017, 20, 55–71. [Google Scholar] [CrossRef]
- Faustino-Rocha, A.; Oliveira, P.A.; Pinho-Oliveira, J.; Teixeira-Guedes, C.; Soares-Maia, R.; da Costa, R.G.; Colaco, B.; Pires, M.J.; Colaco, J.; Ferreira, R.; et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab. Anim. 2013, 42, 217–224. [Google Scholar] [CrossRef]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Larcher, L.M.; Ma, L.; Veedu, R.N. Systematic Screening of Commonly Used Commercial Transfection Reagents towards Efficient Transfection of Single-Stranded Oligonucleotides. Molecules 2018, 23, 2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Rasmussen, M.; Dong, Q.R.; Tepel, M.; Scholze, A. Expression of the NRF2 Target Gene NQO1 Is Enhanced in Mononuclear Cells in Human Chronic Kidney Disease. Oxid. Med. Cell Longev. 2017, 2017, 9091879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olayanju, A.; Copple, I.M.; Bryan, H.K.; Edge, G.T.; Sison, R.L.; Wong, M.W.; Lai, Z.Q.; Lin, Z.X.; Dunn, K.; Sanderson, C.M.; et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic. Biol. Med. 2015, 78, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Kabala-Dzik, A.; Rzepecka-Stojko, A.; Kubina, R.; Jastrzebska-Stojko, Z.; Stojko, R.; Wojtyczka, R.D.; Stojko, J. Migration Rate Inhibition of Breast Cancer Cells Treated by Caffeic Acid and Caffeic Acid Phenethyl Ester: An In Vitro Comparison Study. Nutrients 2017, 9, 1144. [Google Scholar] [CrossRef]
- Yue, P.Y.; Leung, E.P.; Mak, N.K.; Wong, R.N. A simplified method for quantifying cell migration/wound healing in 96-well plates. J. Biomol. Screen 2010, 15, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Atale, N.; Gupta, S.; Yadav, U.C.; Rani, V. Cell-death assessment by fluorescent and nonfluorescent cytosolic and nuclear staining techniques. J. Microsc. 2014, 255, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Fung, A.S.; Jonkman, J.; Tannock, I.F. Quantitative immunohistochemistry for evaluating the distribution of Ki67 and other biomarkers in tumor sections and use of the method to study repopulation in xenografts after treatment with paclitaxel. Neoplasia 2012, 14, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Basilio-de-Oliveira, R.P.; Pannain, V.L. Prognostic angiogenic markers (endoglin, VEGF, CD31) and tumor cell proliferation (Ki67) for gastrointestinal stromal tumors. World J. Gastroenterol. 2015, 21, 6924–6930. [Google Scholar] [CrossRef]
- Onodera, Y.; Motohashi, H.; Takagi, K.; Miki, Y.; Shibahara, Y.; Watanabe, M.; Ishida, T.; Hirakawa, H.; Sasano, H.; Yamamoto, M.; et al. NRF2 immunolocalization in human breast cancer patients as a prognostic factor. Endocr. Relat. Cancer 2014, 21, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Favreau, L.V.; Pickett, C.B. The rat quinone reductase antioxidant response element. Identification of the nucleotide sequence required for basal and inducible activity and detection of antioxidant response element-binding proteins in hepatoma and non-hepatoma cell lines. J. Biol. Chem. 1995, 270, 24468–24474. [Google Scholar] [CrossRef] [Green Version]
- Giudice, A.; Barbieri, A.; Bimonte, S.; Cascella, M.; Cuomo, A.; Crispo, A.; D’Arena, G.; Galdiero, M.; Della Pepa, M.E.; Botti, G.; et al. Dissecting the prevention of estrogen-dependent breast carcinogenesis through Nrf2-dependent and independent mechanisms. Oncol. Targets Ther. 2019, 12, 4937–4953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Boldin-Adamsky, S.; Thimmulappa, R.K.; Rath, S.K.; Ashush, H.; Coulter, J.; Blackford, A.; Goodman, S.N.; Bunz, F.; Watson, W.H.; et al. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 2008, 68, 7975–7984. [Google Scholar] [CrossRef] [Green Version]
- Bialk, P.; Wang, Y.; Banas, K.; Kmiec, E.B. Functional Gene Knockout of NRF2 Increases Chemosensitivity of Human Lung Cancer A549 Cells In Vitro and in a Xenograft Mouse Model. Mol. Ther. Oncolytics 2018, 11, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- Arlt, A.; Sebens, S.; Krebs, S.; Geismann, C.; Grossmann, M.; Kruse, M.L.; Schreiber, S.; Schafer, H. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene 2013, 32, 4825–4835. [Google Scholar] [CrossRef]
- Yang, Y.; Tian, Z.; Guo, R.; Ren, F. Nrf2 Inhibitor, Brusatol in Combination with Trastuzumab Exerts Synergistic Antitumor Activity in HER2-Positive Cancers by Inhibiting Nrf2/HO-1 and HER2-AKT/ERK1/2 Pathways. Oxid. Med. Cell Longev. 2020, 2020, 9867595. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Ye, W.; Huang, C.; Yu, D.; Chen, H.; Deng, T.; Zhang, F.; Lou, B.; Zhang, J.; Shi, K.; et al. Brusatol Enhances the Chemotherapy Efficacy of Gemcitabine in Pancreatic Cancer via the Nrf2 Signalling Pathway. Oxid. Med. Cell Longev. 2018, 2018, 2360427. [Google Scholar] [CrossRef]
- Karathedath, S.; Rajamani, B.M.; Musheer Aalam, S.M.; Abraham, A.; Varatharajan, S.; Krishnamurthy, P.; Mathews, V.; Velayudhan, S.R.; Balasubramanian, P. Role of NF-E2 related factor 2 (Nrf2) on chemotherapy resistance in acute myeloid leukemia (AML) and the effect of pharmacological inhibition of Nrf2. PLoS ONE 2017, 12, e0177227. [Google Scholar] [CrossRef] [Green Version]
- Pouremamali, F.; Farhad, J.; Nasser, S. Nrf2-ME-1 axis is associated with 5-FU resistance in gastric cancer cell line. Process. Biochem. 2020, in press. [Google Scholar] [CrossRef]
- Woo, Y.; Oh, J.; Kim, J.S. Suppression of Nrf2 Activity by Chestnut Leaf Extract Increases Chemosensitivity of Breast Cancer Stem Cells to Paclitaxel. Nutrients 2017, 9, 760. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, J.; Balasubramaniam, J.; Sellamuthu, A.; Ravi, A. An in vitro study on the reversal of epithelial to mesenchymal transition by brusatol and its synergistic properties in triple-negative breast cancer cells. J. Pharm. Pharmacol. 2021, 73, 749–757. [Google Scholar] [CrossRef]
- Yu, X.; Su, X.; Huang, X.; Yao, G.; Song, S. Brusatol: A potential anti-tumor quassinoid from Brucea javanica. Chin. Herb. Med. 2020, 12, 359–366. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Torrente, L.; Sanchez, C.; Moreno, R.; Chowdhry, S.; Cabello, P.; Isono, K.; Koseki, H.; Honda, T.; Hayes, J.D.; Dinkova-Kostova, A.T.; et al. Crosstalk between NRF2 and HIPK2 shapes cytoprotective responses. Oncogene 2017, 36, 6204–6212. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.Z.; Wu, X.H. A small compound spindlactone A sensitizes human endometrial cancer cells to TRAIL-induced apoptosis via the inhibition of NAD(P)H dehydrogenase quinone 1. Oncol. Targets Ther. 2018, 11, 3609–3617. [Google Scholar] [CrossRef] [Green Version]
- Gerard, C.; Goldbeter, A. The balance between cell cycle arrest and cell proliferation: Control by the extracellular matrix and by contact inhibition. Interface Focus 2014, 4, 20130075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, N.M.; Kleeberger, S.R.; Bream, J.H.; Fallon, P.G.; Kensler, T.W.; Yamamoto, M.; Reddy, S.P. Genetic disruption of the Nrf2 compromises cell-cycle progression by impairing GSH-induced redox signaling. Oncogene 2008, 27, 5821–5832. [Google Scholar] [CrossRef] [Green Version]
- Marton, M.; Tihanyi, N.; Gyulavari, P.; Banhegyi, G.; Kapuy, O. NRF2-regulated cell cycle arrest at early stage of oxidative stress response mechanism. PLoS ONE 2018, 13, e0207949. [Google Scholar] [CrossRef] [PubMed]
- Pucci, B.; Kasten, M.; Giordano, A. Cell cycle and apoptosis. Neoplasia 2000, 2, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Niture, S.K.; Kaspar, J.W.; Shen, J.; Jaiswal, A.K. Nrf2 signaling and cell survival. Toxicol Appl. Pharmacol. 2010, 244, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Syu, J.P.; Chi, J.T.; Kung, H.N. Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget 2016, 7, 14659–14672. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.L.; Nasrollahi, S.; Shah, K.N.; Soltisz, A.; Paruchuri, S.; Yun, Y.H.; Luker, G.D.; Bishayee, A.; Tavana, H. Phytochemicals potently inhibit migration of metastatic breast cancer cells. Integr. Biol. 2015, 7, 792–800. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.S.; Zhang, Z.G.; Du, G.Y.; Sun, H.L.; Liu, H.Y.; Zhou, Z.; Gou, X.M.; Wu, X.H.; Yu, X.Y.; Huang, Y.H. Nrf2 promotes breast cancer cell migration via up-regulation of G6PD/HIF-1alpha/Notch1 axis. J. Cell Mol. Med. 2019, 23, 3451–3463. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sno: | Gene Name | Forward Primer | Reverse Primer | Product Size (bp) | Ref. |
|---|---|---|---|---|---|
| Primer sequences used for Homo sapiens | |||||
| 1 | NRF2 | TTCAGCAGCATCCTCTC CACAG | GCATGCTGTTGCTGATACTGG | 139 | [48] |
| 2 | NQO1 | TGCAGCGGCTTTGAAG AAGAAAGG | TCGGCAGGATACTGAAAGTTCGCA | 251 | [49] |
| 3 | β-Actin | TGGATCAGCAAGCAGG AGTATG | GCATTTGCGGTGGACGAT | 57 | [50] |
| 4 | GAPDH | CGACCACTTTGTCAAGC TCA | AGGGGAGATTCAGTGTGGTG | 307 | [51] |
| Primer sequences for mouse EAC cells | |||||
| 5 | NRF2 | TTCTTTCAGCAGCATCC TCTCCAC | ACAGCCTTCAATAGTC CCGTCCAG | 199 | [52] |
| 6 | NQO1 | TATCCTTCCGAGTCATC TCAGC | TCTGCAGCTTCCA GCTTCTTG | 86 | [53] |
| 7 | GAPDH | AGAGAGGGAGGAGGG GAATG | AACAGGGAGGAGCA GAGAGCAC | 200 | [54] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bovilla, V.R.; Kuruburu, M.G.; Bettada, V.G.; Krishnamurthy, J.; Sukocheva, O.A.; Thimmulappa, R.K.; Shivananju, N.S.; Balakrishna, J.P.; Madhunapantula, S.V. Targeted Inhibition of Anti-Inflammatory Regulator Nrf2 Results in Breast Cancer Retardation In Vitro and In Vivo. Biomedicines 2021, 9, 1119. https://doi.org/10.3390/biomedicines9091119
Bovilla VR, Kuruburu MG, Bettada VG, Krishnamurthy J, Sukocheva OA, Thimmulappa RK, Shivananju NS, Balakrishna JP, Madhunapantula SV. Targeted Inhibition of Anti-Inflammatory Regulator Nrf2 Results in Breast Cancer Retardation In Vitro and In Vivo. Biomedicines. 2021; 9(9):1119. https://doi.org/10.3390/biomedicines9091119
Chicago/Turabian StyleBovilla, Venugopal R., Mahadevaswamy G. Kuruburu, Vidya G. Bettada, Jayashree Krishnamurthy, Olga A. Sukocheva, Rajesh K. Thimmulappa, Nanjunda Swamy Shivananju, Janardhan P. Balakrishna, and SubbaRao V. Madhunapantula. 2021. "Targeted Inhibition of Anti-Inflammatory Regulator Nrf2 Results in Breast Cancer Retardation In Vitro and In Vivo" Biomedicines 9, no. 9: 1119. https://doi.org/10.3390/biomedicines9091119