
Unraveling the Complexity of HDL Remodeling: On the Hunt to Restore HDL Quality



Abstract
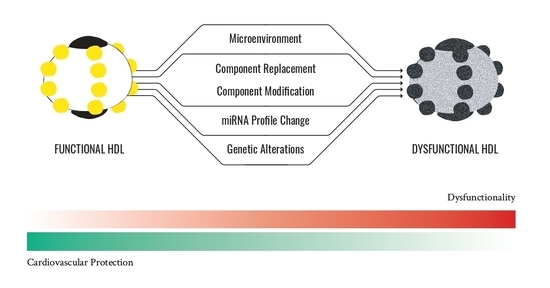
:
1. The HDL-C Hypothesis Revisited
2. HDL Particle Alterations: On the Lookout for What Makes HDL Particles Lose Their Protective Functions
2.1. HDL Component Modifications
2.1.1. Oxidation
2.1.2. Carbamylation
2.1.3. Glycation
2.2. HDL Component Replacements
2.3. HDL miRNA Profile
2.4. Genetic Alterations Affecting HDL
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Castelli, W.P.; Garrison, R.J.; Wilson, P.W.F.; Abbott, R.D.; Kalousdian, S.; Kannel, W.B. Incidence of Coronary Heart Disease and Lipoprotein Cholesterol Levels. JAMA 1986, 256, 2835. [Google Scholar] [CrossRef]
- Stampfer, M.J.; Sacks, F.M.; Salvini, S.; Willett, W.C.; Hennekens, C.H. A Prospective Study of Cholesterol, Apolipoproteins, and the Risk of Myocardial Infarction. N. Engl. J. Med. 1991, 325, 373–381. [Google Scholar] [CrossRef] [PubMed]
- The Emerging Risk Factors Collaboration. Major Lipids, Apolipoproteins, and Risk of Vascular Disease. JAMA 2009, 302, 1993. [Google Scholar] [CrossRef] [Green Version]
- Mackey, R.H.; Greenland, P.; Goff, D.C.; Lloyd-Jones, D.; Sibley, C.T.; Mora, S.; Mora, S. High-Density Lipoprotein Cholesterol and Particle Concentrations, Carotid Atherosclerosis, and Coronary Events: MESA (Multi-Ethnic Study of Atherosclerosis). J. Am. Coll. Cardiol. 2012, 60, 508–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergeer, M.; Holleboom, A.G.; Kastelein, J.J.P.; Kuivenhoven, J.A. The HDL Hypothesis: Does High-Density Lipoprotein Protect from Atherosclerosis? J. Lipid Res. 2010, 51, 2058–2073. [Google Scholar] [CrossRef] [Green Version]
- Castelli, W.P.; Doyle, J.T.; Gordon, T.; Hames, C.G.; Hjortland, M.C.; Hulley, S.B.; Kagan, A.; Zukel, W.J. HDL Cholesterol and Other Lipids in Coronary Heart Disease. The Cooperative Lipoprotein Phenotyping Study. Circulation 1977, 55, 767–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport. Circ. Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef]
- Badimon, J.J.; Badimon, L.; Fuster, V. Regression of Atherosclerotic Lesions by High Density Lipoprotein Plasma Fraction in the Cholesterol-Fed Rabbit. J. Clin. Investig. 1990, 85, 1234–1241. [Google Scholar] [CrossRef] [Green Version]
- Badimon, J.; Badimon, L.; Galvez, A.; Dische, R.; Fuster, V. High Density Lipoprotein Plasma Fractions Inhibit Aortic Fatty Streaks in Cholesterol-Fed Rabbits. Lab Investig. 1989, 60, 455–461. [Google Scholar]
- Brites, F.; Martin, M.; Guillas, I.; Kontush, A. Antioxidative Activity of High-Density Lipoprotein (HDL): Mechanistic Insights into Potential Clinical Benefit. BBA Clin. 2017, 8, 66–77. [Google Scholar] [CrossRef]
- Riwanto, M.; Landmesser, U. High Density Lipoproteins and Endothelial Functions: Mechanistic Insights and Alterations in Cardiovascular Disease. J. Lipid Res. 2013, 54, 3227–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moya, C.; Máñez, S. Paraoxonases: Metabolic Role and Pharmacological Projection. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 349–359. [Google Scholar] [CrossRef]
- Keul, P.; Polzin, A.; Kaiser, K.; Gräler, M.; Dannenberg, L.; Daum, G.; Heusch, G.; Levkau, B. Potent Anti-Inflammatory Properties of HDL in Vascular Smooth Muscle Cells Mediated by HDL-S1P and Their Impairment in Coronary Artery Disease Due to Lower HDL-S1P: A New Aspect of HDL Dysfunction and Its Therapy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 1482–1495. [Google Scholar] [CrossRef] [PubMed]
- Van der Stoep, M.; Korporaal, S.J.A.; van Eck, M. High-Density Lipoprotein as a Modulator of Platelet and Coagulation Responses. Cardiovasc. Res. 2014, 103, 362–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, D.W.; Chen, J.; Ling, M.; Fu, X.; Blevins, T.; Parsons, S.; Le, J.; Harris, J.; Martin, T.R.; Konkle, B.A.; et al. High-Density Lipoprotein Modulates Thrombosis by Preventing von Willebrand Factor Self-Association and Subsequent Platelet Adhesion. Blood 2016, 127, 637–645. [Google Scholar] [CrossRef]
- Miller, N.E.; Chigaev, A.; Khan, H.; Khan, A.W.; Mardan, U.; Colantuoni, A.; Franceschini, G.; Gomaraschi, M.; Calabresi, L. Protective Effects of HDL Against Ischemia/Reperfusion Injury. Front. Pharmacol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Schwertani, A.; Choi, H.Y.; Genest, J. HDLs and the Pathogenesis of Atherosclerosis. Curr. Opin. Cardiol. 2018, 33, 311–316. [Google Scholar] [CrossRef]
- Robert, J.; Osto, E.; von Eckardstein, A. The Endothelium Is Both a Target and a Barrier of HDL’s Protective Functions. Cells 2021, 10, 1041. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G. LDL-Cholesterol versus HDL-Cholesterol in the Atherosclerotic Plaque: Inflammatory Resolution versus Thrombotic Chaos. Ann. N. Y. Acad. Sci. 2012, 1254, 18–32. [Google Scholar] [CrossRef]
- Rubin, E.M.; Krauss, R.M.; Spangler, E.A.; Verstuyft, J.G.; Clift, S.M. Inhibition of Early Atherogenesis in Transgenic Mice by Human Apolipoprotein AI. Nature 1991, 353, 265–267. [Google Scholar] [CrossRef]
- Méndez-Lara, K.A.; Farré, N.; Santos, D.; Rivas-Urbina, A.; Metso, J.; Sánchez-Quesada, J.L.; Llorente-Cortes, V.; Errico, T.L.; Lerma, E.; Jauhiainen, M.; et al. Human ApoA-I Overexpression Enhances Macrophage-Specific Reverse Cholesterol Transport but Fails to Prevent Inherited Diabesity in Mice. Int. J. Mol. Sci. 2019, 20, 655. [Google Scholar] [CrossRef] [Green Version]
- Duverger, N.; Kruth, H.; Emmanuel, F.; Caillaud, J.-M.; Viglietta, C.; Castro, G.; Tailleux, A.; Fievet, C.; Fruchart, J.C.; Houdebine, L.M.; et al. Inhibition of Atherosclerosis Development in Cholesterol-Fed Human Apolipoprotein A-I–Transgenic Rabbits. Circulation 1996, 94, 713–717. [Google Scholar] [CrossRef]
- Ibanez, B.; Giannarelli, C.; Cimmino, G.; Santos-Gallego, C.G.; Alique, M.; Pinero, A.; Vilahur, G.; Fuster, V.; Badimon, L.; Badimon, J.J. Recombinant HDL(Milano) Exerts Greater Anti-Inflammatory and Plaque Stabilizing Properties than HDL(Wild-Type). Atherosclerosis 2012, 220, 72–77. [Google Scholar] [CrossRef]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Hólm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL Cholesterol and Risk of Myocardial Infarction: A Mendelian Randomisation Study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.P.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.-C.; Waters, D.D.; et al. Effects of Torcetrapib in Patients at High Risk for Coronary Events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of Dalcetrapib in Patients with a Recent Acute Coronary Syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [Green Version]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef]
- Group, T.H.C. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef]
- Keene, D.; Price, C.; Shun-Shin, M.J.; Francis, D.P. Effect on Cardiovascular Risk of High Density Lipoprotein Targeted Drug Treatments Niacin, Fibrates, and CETP Inhibitors: Meta-Analysis of Randomised Controlled Trials Including 117,411 Patients. BMJ 2014, 349, g4379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Extreme High High-Density Lipoprotein Cholesterol Is Paradoxically Associated with High Mortality in Men and Women: Two Prospective Cohort Studies. Eur. Heart J. 2017, 38, 2478–2486. [Google Scholar] [CrossRef] [Green Version]
- Dron, J.S.; Wang, J.; Low-Kam, C.; Khetarpal, S.A.; Robinson, J.F.; McIntyre, A.D.; Ban, M.R.; Cao, H.; Rhainds, D.; Dubé, M.-P.; et al. Polygenic Determinants in Extremes of High-Density Lipoprotein Cholesterol. J. Lipid Res. 2017, 58, 2162–2170. [Google Scholar] [CrossRef] [Green Version]
- Rosenson, R.S.; Brewer, H.B.; Barter, P.J.; Björkegren, J.L.M.; Chapman, M.J.; Gaudet, D.; Kim, D.S.; Niesor, E.; Rye, K.-A.; Sacks, F.M.; et al. HDL and Atherosclerotic Cardiovascular Disease: Genetic Insights into Complex Biology. Nat. Rev. Cardiol. 2018, 15, 9–19. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G. HDL Particles—More Complex than We Thought. Thromb. Haemost. 2014, 112, 857. [Google Scholar] [CrossRef]
- Jensen, J.; Miedema, M.D. Quality Over Quantity: The Role of HDL Cholesterol Efflux Capacity in Atherosclerotic Cardiovascular Disease—American College of Cardiology. J. Am. Coll. Cardiol. 2017, 69, 246–247. [Google Scholar]
- Galvani, S.; Hla, T. Quality vs. Quantity: Making HDL Great Again. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1018–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilahur, G.; Cubedo, J.; Padró, T.; Casaní, L.; Mendieta, G.; González, A.; Badimon, L. Intake of Cooked Tomato Sauce Preserves Coronary Endothelial Function and Improves Apolipoprotein A-I and Apolipoprotein J Protein Profile in High-Density Lipoproteins. Transl. Res. 2015, 166, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.M.; Shishehbor, M.H.; Ansell, B.J. High-Density Lipoprotein as a Therapeutic Target. JAMA 2007, 298, 786. [Google Scholar] [CrossRef]
- Ruiz-Ramie, J.J.; Barber, J.L.; Sarzynski, M.A. Effects of Exercise on HDL Functionality. Curr. Opin. Lipidol. 2019, 30, 16–23. [Google Scholar] [CrossRef]
- Nystoriak, M.A.; Bhatnagar, A. Cardiovascular Effects and Benefits of Exercise. Front. Cardiovasc. Med. 2018, 5, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardagjy, A.S.; Steinberg, F.M. Relationship between HDL Functional Characteristics and Cardiovascular Health and Potential Impact of Dietary Patterns: A Narrative Review. Nutrients 2019, 11, 1231. [Google Scholar] [CrossRef] [Green Version]
- Grao-Cruces, E.; Varela, L.M.; Martin, M.E.; Bermudez, B.; Montserrat-de la Paz, S. High-Density Lipoproteins and Mediterranean Diet: A Systematic Review. Nutrients 2021, 13, 955. [Google Scholar] [CrossRef]
- Dattilo, A.; Kris-Etherton, P. Effects of Weight Reduction on Blood Lipids and Lipoproteins: A Meta-Analysis. Am. J. Clin. Nutr. 1992, 56. [Google Scholar] [CrossRef]
- Zomer, E.; Gurusamy, K.; Leach, R.; Trimmer, C.; Lobstein, T.; Morris, S.; James, W.P.T.; Finer, N. Interventions That Cause Weight Loss and the Impact on Cardiovascular Risk Factors: A Systematic Review and Meta-Analysis. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2016, 17, 1001–1011. [Google Scholar] [CrossRef]
- Hasan, B.; Nayfeh, T.; Alzuabi, M.; Wang, Z.; Kuchkuntla, A.R.; Prokop, L.J.; Newman, C.B.; Murad, M.H.; Rajjo, T.I. Weight Loss and Serum Lipids in Overweight and Obese Adults: A Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef] [PubMed]
- Christou, G.A.; Kiortsis, D.N. Adiponectin and Lipoprotein Metabolism. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2013, 14, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Ronsein, G.E.; Vaisar, T. Deepening Our Understanding of HDL Proteome. Expert Rev. Proteom. 2019, 16, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Kajani, S.; Curley, S.; McGillicuddy, F.C. Unravelling HDL-Looking beyond the Cholesterol Surface to the Quality Within. Int. J. Mol. Sci. 2018, 19, 1971. [Google Scholar] [CrossRef] [Green Version]
- Ben-Aicha, S.; Badimon, L.; Vilahur, G. Advances in HDL: Much More than Lipid Transporters. Int. J. Mol. Sci. 2020, 21, 732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.J.; Chen, Q.; Zabalawi, M.; Anderson, R.; Wilson, M.; Weinberg, R.; Sorci-Thomas, M.G.; Rudel, L.L. Is the Oxidation of High-Density Lipoprotein Lipids Different Than the Oxidation of Low-Density Lipoprotein Lipids? Biochemistry 2001. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.Z.; Kar, N.S.; Chen, X.; Choi, J.; Salomon, R.G.; Febbraio, M.; Podrez, E.A. Specific Oxidized Phospholipids Inhibit Scavenger Receptor Bi-Mediated Selective Uptake of Cholesteryl Esters. J. Biol. Chem. 2008, 283, 10408–10414. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Podrez, E.A. Characterization of Covalent Modifications of HDL Apoproteins by Endogenous Oxidized Phospholipids. Free Radic. Biol. Med. 2018, 115, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Martínez-López, D.; Camafeita, E.; Cedó, L.; Roldan-Montero, R.; Jorge, I.; García-Marqués, F.; Gómez-Serrano, M.; Bonzon-Kulichenko, E.; Blanco-Vaca, F.; Blanco-Colio, L.M.; et al. APOA1 Oxidation Is Associated to Dysfunctional High-Density Lipoproteins in Human Abdominal Aortic Aneurysm. EBioMedicine 2019, 43, 43–53. [Google Scholar] [CrossRef]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a Catalyst for Lipoprotein Oxidation, Is Expressed in Human Atherosclerotic Lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, A.; Chan, G.K.L.; Boatz, J.C.; Li, N.J.; Inoue, A.P.; Wong, J.C.; van der Wel, P.C.A.; Cavigiolio, G. Methionine Oxidized Apolipoprotein A-I at the Crossroads of HDL Biogenesis and Amyloid Formation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 3149–3165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkowski, A.; Carta, S.; Lu, R.; Yokoyama, S.; Rubartelli, A.; Cavigiolio, G. Oxidation of Methionine Residues in Human Apolipoprotein A-I Generates a Potent pro-Inflammatory Molecule. J. Biol. Chem. 2019, 294, 3634–3646. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.; Bergt, C.; Fu, X.; Green, P.; Voss, J.C.; Oda, M.N.; Oram, J.F.; Heinecke, J.W. Tyrosine 192 in Apolipoprotein A-I Is the Major Site of Nitration and Chlorination by Myeloperoxidase, but Only Chlorination Markedly Impairs ABCA1-Dependent Cholesterol Transport. J. Biol. Chem. 2005, 280, 5983–5993. [Google Scholar] [CrossRef] [Green Version]
- Hewing, B.; Parathath, S.; Barrett, T.; Chung, W.K.K.; Astudillo, Y.M.; Hamada, T.; Ramkhelawon, B.; Tallant, T.C.; Yusufishaq, M.S.S.; Didonato, J.A.; et al. Effects of Native and Myeloperoxidase-Modified Apolipoprotein a-I on Reverse Cholesterol Transport and Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 779–789. [Google Scholar] [CrossRef]
- Jin, Z.; Zhou, L.; Tian, R.; Lu, N. Myeloperoxidase Targets Apolipoprotein A-I for Site-Specific Tyrosine Chlorination in Atherosclerotic Lesions and Generates Dysfunctional High-Density Lipoprotein. Chem. Res. Toxicol. 2021. [Google Scholar] [CrossRef]
- Zhou, B.; Zu, L.; Chen, Y.; Zheng, X.; Wang, Y.; Pan, B.; Dong, M.; Zhou, E.; Zhao, M.; Zhang, Y.; et al. Myeloperoxidase-Oxidized High Density Lipoprotein Impairs Atherosclerotic Plaque Stability by Inhibiting Smooth Muscle Cell Migration. Lipids Health Dis. 2017, 16, 3. [Google Scholar] [CrossRef] [Green Version]
- Zamanian-Daryoush, M.; Gogonea, V.; DiDonato, A.J.; Buffa, J.A.; Choucair, I.; Levison, B.S.; Hughes, R.A.; Ellington, A.D.; Huang, Y.; Li, X.S.; et al. Site-Specific 5-Hydroxytryptophan Incorporation into Apolipoprotein A-I Impairs Cholesterol Efflux Activity and High-Density Lipoprotein Biogenesis. J. Biol. Chem. 2020, 295, 4836–4848. [Google Scholar] [CrossRef]
- Huang, Y.; DiDonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An Abundant Dysfunctional Apolipoprotein A1 in Human Atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Holzer, M.; Gauster, M.; Pfeifer, T.; Wadsack, C.; Fauler, G.; Stiegler, P.; Koefeler, H.; Beubler, E.; Schuligoi, R.; Heinemann, A.; et al. Protein Carbamylation Renders High-Density Lipoprotein Dysfunctional. Antioxid. Redox Signal. 2011, 14, 2337–2346. [Google Scholar] [CrossRef] [Green Version]
- Holzer, M.; Zangger, K.; El-Gamal, D.; Binder, V.; Curcic, S.; Konya, V.; Schuligoi, R.; Heinemann, A.; Marsche, G. Myeloperoxidase-Derived Chlorinating Species Induce Protein Carbamylation through Decomposition of Thiocyanate and Urea: Novel Pathways Generating Dysfunctional High-Density Lipoprotein. Antioxid. Redox Signal. 2012, 17, 1043–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Nicholls, S.J.; Rodriguez, E.R.; Kummu, O.; Hörkkö, S.; Barnard, J.; Reynolds, W.F.; Topol, E.J.; DiDonato, J.A.; Hazen, S.L. Protein Carbamylation Links Inflammation, Smoking, Uremia and Atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef]
- Matsunaga, T.; Nakajima, T.; Miyazaki, T.; Koyama, I.; Hokari, S.; Inoue, I.; Kawai, S.; Shimomura, H.; Katayama, S.; Hara, A.; et al. Glycated High-Density Lipoprotein Regulates Reactive Oxygen Species and Reactive Nitrogen Species in Endothelial Cells. Metab. Clin. Exp. 2003, 52, 42–49. [Google Scholar] [CrossRef]
- Cochran, B.J.; Ong, K.L.; Manandhar, B.; Rye, K.A. High Density Lipoproteins and Diabetes. Cells 2021, 10, 850. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Qian, M.-M.; Liu, P.-L.; Zhang, L.; Wang, Y.; Liu, D.-H. Glycation of High-Density Lipoprotein Triggers Oxidative Stress and Promotes the Proliferation and Migration of Vascular Smooth Muscle Cells. J. Geriatr. Cardiol. JGC 2017, 14, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Nobecourt, E.; Davies, M.J.; Brown, B.E.; Curtiss, L.K.; Bonnet, D.J.; Charlton, F.; Januszewski, A.S.; Jenkins, A.J.; Barter, P.J.; Rye, K.-A. The Impact of Glycation on Apolipoprotein A-I Structure and Its Ability to Activate Lecithin:Cholesterol Acyltransferase. Diabetologia 2007, 50, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Femlak, M.; Gluba-Brzózka, A.; Ciałkowska-Rysz, A.; Rysz, J. The Role and Function of HDL in Patients with Diabetes Mellitus and the Related Cardiovascular Risk. Lipids Health Dis. 2017, 16, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, A.; Murphy, A.J.; Coughlan, M.T.; Thomas, M.C.; Forbes, J.M.; O’Brien, R.; Cooper, M.E.; Chin-Dusting, J.P.F.; Sviridov, D. Advanced Glycation of Apolipoprotein A-I Impairs Its Anti-Atherogenic Properties. Diabetologia 2007, 50, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.J.; Lu, L.; Zhang, R.Y.; Du, R.; Shen, Y.; Zhang, Q.; Yang, Z.K.; Chen, Q.J.; Shen, W.F. Glycation of Apoprotein A-I Is Associated with Coronary Artery Plaque Progression in Type 2 Diabetic Patients. Diabetes Care 2013, 36, 1312–1320. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Ding, F.H.; Sun, J.T.; Pu, L.J.; Zhang, R.Y.; Zhang, Q.; Chen, Q.J.; Shen, W.F.; Lu, L. Association of Elevated ApoA-I Glycation and Reduced HDL-Associated Paraoxonase1, 3 Activity, and Their Interaction with Angiographic Severity of Coronary Artery Disease in Patients with Type 2 Diabetes Mellitus. Cardiovasc. Diabetol. 2015, 14, 52. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, S.R.; Osme, A.; Ilchenko, S.; Golizeh, M.; Lee, K.; Wang, S.; Bena, J.; Previs, S.F.; Smith, J.D.; Kasumov, T. Glycation Reduces the Stability of ApoAI and Increases HDL Dysfunction in Diet-Controlled Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 388–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lê, Q.H.; El Alaoui, M.; Véricel, E.; Ségrestin, B.; Soulère, L.; Guichardant, M.; Lagarde, M.; Moulin, P.; Calzada, C. Glycoxidized HDL, HDL Enriched with Oxidized Phospholipids and HDL from Diabetic Patients Inhibit Platelet Function. J. Clin. Endocrinol. Metab. 2015, 100, 2006–2014. [Google Scholar] [CrossRef] [Green Version]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the Complexities of the HDL Lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef] [Green Version]
- Waldie, S.; Sebastiani, F.; Browning, K.; Maric, S.; Lind, T.K.; Yepuri, N.; Darwish, T.A.; Moulin, M.; Strohmeier, G.; Pichler, H.; et al. Lipoprotein Ability to Exchange and Remove Lipids from Model Membranes as a Function of Fatty Acid Saturation and Presence of Cholesterol. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2020, 1865, 158769. [Google Scholar] [CrossRef]
- Chiesa, S.T.; Charakida, M. High-Density Lipoprotein Function and Dysfunction in Health and Disease. Cardiovasc. Drugs Ther. 2019, 33, 207. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; de Beer, M.C.; de Beer, F.C.; van der Westhuyzen, D.R. Serum Amyloid A Is a Ligand for Scavenger Receptor Class B Type I and Inhibits High Density Lipoprotein Binding and Selective Lipid Uptake. J. Biol. Chem. 2005, 280, 2954–2961. [Google Scholar] [CrossRef] [Green Version]
- Schuchardt, M.; Prüfer, N.; Tu, Y.; Herrmann, J.; Hu, X.-P.; Chebli, S.; Dahlke, K.; Zidek, W.; van der Giet, M.; Tölle, M. Dysfunctional High-Density Lipoprotein Activates Toll-like Receptors via Serum Amyloid A in Vascular Smooth Muscle Cells. Sci. Rep. 2019, 9, 3421. [Google Scholar] [CrossRef] [Green Version]
- Han, C.Y.; Tang, C.; Guevara, M.E.; Wei, H.; Wietecha, T.; Shao, B.; Subramanian, S.; Omer, M.; Wang, S.; O’Brien, K.D.; et al. Serum Amyloid A Impairs the Antiinflammatory Properties of HDL. J. Clin. Investig. 2016, 126, 266–281. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.; Pennathur, S.; Heinecke, J.W. Myeloperoxidase Targets Apolipoprotein A-I, the Major High Density Lipoprotein Protein, for Site-Specific Oxidation in Human Atherosclerotic Lesions. J. Biol. Chem. 2012, 287, 6375–6386. [Google Scholar] [CrossRef] [Green Version]
- Van Lenten, B.J.; Hama, S.Y.; de Beer, F.C.; Stafforini, D.M.; McIntyre, T.M.; Prescott, S.M.; la Du, B.N.; Fogelman, A.M.; Navab, M. Anti-Inflammatory HDL Becomes pro-Inflammatory during the Acute Phase Response. Loss of Protective Effect of HDL against LDL Oxidation in Aortic Wall Cell Cocultures. J. Clin. Investig. 1995, 96, 2758–2767. [Google Scholar] [CrossRef]
- White, R.; Giordano, S.; Datta, G. Role of HDL-Associated Proteins and Lipids in the Regulation of Inflammation. In Advances in Lipoprotein Research; InTech: London, UK, 2017; ISBN 978-953-51-3048-2. [Google Scholar]
- Van Lenten, B.J.; Wagner, A.C.; Nayak, D.P.; Hama, S.; Navab, M.; Fogelman, A.M. High-Density Lipoprotein Loses Its Anti-Inflammatory Properties During Acute Influenza A Infection. Circulation 2001, 103, 2283–2288. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, S.D.; Sok, D.-E. Preferable Stimulation of PON1 Arylesterase Activity by Phosphatidylcholines with Unsaturated Acyl Chains or Oxidized Acyl Chains at Sn-2 Position. Biochim. Biophys. Acta (BBA) Biomembr. 2006, 1758, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.J.; Sorci-Thomas, M.G. SAA: A Link between Cholesterol Efflux Capacity and Inflammation? J. Lipid Res. 2015, 56, 1383–1385. [Google Scholar] [CrossRef] [Green Version]
- Bergmeier, C.; Siekmeier, R.; Gross, W. Distribution Spectrum of Paraoxonase Activity in HDL Fractions. Clin. Chem. 2004, 50, 2309–2315. [Google Scholar] [CrossRef]
- Zewinger, S.; Drechsler, C.; Kleber, M.E.; Dressel, A.; Riffel, J.; Triem, S.; Lehmann, M.; Kopecky, C.; Säemann, M.D.; Lepper, P.M.; et al. Serum Amyloid A: High-Density Lipoproteins Interaction and Cardiovascular Risk. Eur. Heart J. 2015, 36, ehv352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zewinger, S.; Kleber, M.E.; Rohrer, L.; Lehmann, M.; Triem, S.; Jennings, R.T.; Petrakis, I.; Dressel, A.; Lepper, P.M.; Scharnagl, H.; et al. Symmetric Dimethylarginine, High-Density Lipoproteins and Cardiovascular Disease. Eur. Heart J. 2017, 38, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, F.A.H.; de Oliveira Izar, M.C. High-Sensitivity C-Reactive Protein and Cardiovascular Disease across Countries and Ethnicities. Clinics 2016, 71, 235–242. [Google Scholar] [CrossRef]
- Van Capelleveen, J.C.; Bernelot Moens, S.J.; Yang, X.; Kastelein, J.J.P.; Wareham, N.J.; Zwinderman, A.H.; Stroes, E.S.G.; Witztum, J.L.; Hovingh, G.K.; Khaw, K.-T.; et al. Apolipoprotein C-III Levels and Incident Coronary Artery Disease Risk: The EPIC-Norfolk Prospective Population Study. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1206–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilahur, G.; Gutiérrez, M.; Casaní, L.; Cubedo, J.; Capdevila, A.; Pons-Llado, G.; Carreras, F.; Hidalgo, A.; Badimon, L. Hypercholesterolemia Abolishes High-Density Lipoprotein-Related Cardioprotective Effects in the Setting of Myocardial Infarction. J. Am. Coll. Cardiol. 2015, 66, 2469–2470. [Google Scholar] [CrossRef] [Green Version]
- Padró, T.; Cubedo, J.; Camino, S.; Béjar, M.T.; Ben-Aicha, S.; Mendieta, G.; Escolà-Gil, J.C.; Escate, R.; Gutiérrez, M.; Casani, L.; et al. Detrimental Effect of Hypercholesterolemia on High-Density Lipoprotein Particle Remodeling in Pigs. J. Am. Coll. Cardiol. 2017, 70, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Hafiane, A.; Jabor, B.; Ruel, I.; Ling, J.; Genest, J. High-Density Lipoprotein Mediated Cellular Cholesterol Efflux in Acute Coronary Syndromes. Am. J. Cardiol. 2014, 113, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Cardner, M.; Yalcinkaya, M.; Goetze, S.; Luca, E.; Balaz, M.; Hunjadi, M.; Hartung, J.; Shemet, A.; Kränkel, N.; Radosavljevic, S.; et al. Structure-Function Relationships of HDL in Diabetes and Coronary Heart Disease. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilahur, G.; Ben-Aicha, S.; Gutierrez, M.; Mendieta, G.; Casani, L.; Carreras, F.; Hidalgo, A.B.L. Las HDL Pierden Su Efecto Protector En Hipercolesterolemia; Sociedad Española de Cardiología: Madrid, Spain, 2017; Volume 70. [Google Scholar]
- Abu-Assi, E.; López-López, A.; González-Salvado, V.; Redondo-Diéguez, A.; Peña-Gil, C.; Bouzas-Cruz, N.; Raposeiras-Roubín, S.; Riziq-Yousef Abumuaileq, R.; García-Acuña, J.M.; González-Juanatey, J.R. The Risk of Cardiovascular Events After an Acute Coronary Event Remains High, Especially During the First Year, Despite Revascularization. Rev. Española Cardiol. 2016, 69, 11–18. [Google Scholar] [CrossRef]
- Ben-Aicha, S.; Casaní, L.; Muñoz-García, N.; Joan-Babot, O.; Peña, E.; Aržanauskaitė, M.; Gutierrez, M.; Mendieta, G.; Padró, T.; Badimon, L.; et al. HDL (High-Density Lipoprotein) Remodeling and Magnetic Resonance Imaging-Assessed Atherosclerotic Plaque Burden: Study in a Preclinical Experimental Model. Arterioscler. Thromb. Vasc. Biol. 2020. [Google Scholar] [CrossRef]
- Sacks, F.M.; Liang, L.; Furtado, J.D.; Cai, T.; Davidson, W.S.; He, Z.; McClelland, R.L.; Rimm, E.B.; Jensen, M.K. Protein-Defined Subspecies of HDLs (High-Density Lipoproteins) and Differential Risk of Coronary Heart Disease in 4 Prospective Studies. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2714–2727. [Google Scholar] [CrossRef] [PubMed]
- Vaisar, T.; Pennathur, S.; Green, P.S.; Gharib, S.A.; Hoofnagle, A.N.; Cheung, M.C.; Byun, J.; Vuletic, S.; Kassim, S.; Singh, P.; et al. Shotgun Proteomics Implicates Protease Inhibition and Complement Activation in the Antiinflammatory Properties of HDL. J. Clin. Investig. 2007, 117, 746–756. [Google Scholar] [CrossRef]
- Malajczuk, C.; Gandhi, N.; Mancera, R. Structure and Intermolecular Interactions in Spheroidal High-Density Lipoprotein Subpopulations. J. Struct. Biol. X 2020, 5. [Google Scholar] [CrossRef]
- Karathanasis, S.; Freeman, L.; Gordon, S.; Remaley, A.T. The Changing Face of HDL and the Best Way to Measure It. Clin. Chem. 2017, 63. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.S.; Khokhar, A.A.; May, H.T.; Kulkarni, K.R.; Blaha, M.J.; Joshi, P.H.; Toth, P.P.; Muhlestein, J.B.; Anderson, J.L.; Knight, S.; et al. HDL Cholesterol Subclasses, Myocardial Infarction, and Mortality in Secondary Prevention: The Lipoprotein Investigators Collaborative. Eur. Heart J. 2015, 36, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Cubedo, J.; Padró, T.; García-Arguinzonis, M.; Vilahur, G.; Miñambres, I.; Pou, J.M.; Ybarra, J.; Badimon, L. A Novel Truncated Form of Apolipoprotein A-I Transported by Dense LDL Is Increased in Diabetic Patients. J. Lipid Res. 2015, 56, 1762–1773. [Google Scholar] [CrossRef] [Green Version]
- Ben-Aicha, S.; Escate, R.; Casaní, L.; Padró, T.; Peña, E.; Arderiu, G.; Mendieta, G.; Badimón, L.; Vilahur, G. High-Density Lipoprotein Remodelled in Hypercholesterolaemic Blood Induce Epigenetically Driven down-Regulation of Endothelial HIF-1α Expression in a Preclinical Animal Model. Cardiovasc. Res. 2020, 116, 1288–1299. [Google Scholar] [CrossRef] [Green Version]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs Are Transported in Plasma and Delivered to Recipient Cells by High-Density Lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, J.; Riwanto, M.; Besler, C.; Knau, A.; Fichtlscherer, S.; Röxe, T.; Zeiher, A.M.; Landmesser, U.; Dimmeler, S. Characterization of Levels and Cellular Transfer of Circulating Lipoprotein-Bound MicroRNAs. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1392–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niculescu, L.S.; Simionescu, N.; Sanda, G.M.; Carnuta, M.G.; Stancu, C.S.; Popescu, A.C.; Popescu, M.R.; Vlad, A.; Dimulescu, D.R.; Simionescu, M.; et al. MiR-486 and MiR-92a Identified in Circulating HDL Discriminate between Stable and Vulnerable Coronary Artery Disease Patients. PLoS ONE 2015, 10, e0140958. [Google Scholar] [CrossRef] [PubMed]
- Scicali, R.; di Pino, A.; Pavanello, C.; Ossoli, A.; Strazzella, A.; Alberti, A.; di Mauro, S.; Scamporrino, A.; Urbano, F.; Filippello, A.; et al. Analysis of HDL-MicroRNA Panel in Heterozygous Familial Hypercholesterolemia Subjects with LDL Receptor Null or Defective Mutation. Sci. Rep. 2019, 9, 20354. [Google Scholar] [CrossRef] [Green Version]
- HGMD® Gene Result. Available online: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=APOA1 (accessed on 26 May 2020).
- Tall, A.R. Plasma High Density Lipoproteins. Metabolism and Relationship to Atherogenesis. J. Clin. Investig. 1990, 86, 379–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirtori, C.R.; Calabresi, L.; Franceschini, G.; Baldassarre, D.; Amato, M.; Johansson, J.; Salvetti, M.; Monteduro, C.; Zulli, R.; Muiesan, M.L.; et al. Cardiovascular Status of Carriers of the Apolipoprotein A-I Milano Mutant. Circulation 2001, 103, 1949–1954. [Google Scholar] [CrossRef] [Green Version]
- Arciello, A.; Piccoli, R.; Monti, D.M. Apolipoprotein A-I: The Dual Face of a Protein. FEBS Lett. 2016, 590, 4171–4179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase, C.L.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Population-Based Resequencing of APOA1 in 10,330 Individuals: Spectrum of Genetic Variation, Phenotype, and Comparison with Extreme Phenotype Approach. PLoS Genet. 2012, 8, e1003063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, A.J.; Hegele, R.A.; Burnett, J.R. Tangier Disease. Curr. Opin. Lipidol. 2020, 31, 80–84. [Google Scholar] [CrossRef]
- Tietjen, I.; Hovingh, G.K.; Singaraja, R.R.; Radomski, C.; Barhdadi, A.; McEwen, J.; Chan, E.; Mattice, M.; Legendre, A.; Franchini, P.L.; et al. Segregation of LIPG, CETP, and GALNT2 Mutations in Caucasian Families with Extremely High HDL Cholesterol. PLoS ONE 2012, 7, e37437. [Google Scholar] [CrossRef] [Green Version]
- Brunham, L.R.; Tietjen, I.; Bochem, A.E.; Singaraja, R.R.; Franchini, P.L.; Radomski, C.; Mattice, M.; Legendre, A.; Hovingh, G.K.; Kastelein, J.J.P.; et al. Novel Mutations in Scavenger Receptor BI Associated with High HDL Cholesterol in Humans. Clin. Genet. 2011, 79, 575–581. [Google Scholar] [CrossRef]
- Asztalos, B.F.; Schaefer, E.J.; Horvath, K.V.; Yamashita, S.; Miller, M.; Franceschini, G.; Calabresi, L. Role of LCAT in HDL Remodeling: Investigation of LCAT Deficiency States. J. Lipid Res. 2007, 48, 592–599. [Google Scholar] [CrossRef] [Green Version]
- Tietjen, I.; Hovingh, G.K.; Singaraja, R.; Radomski, C.; McEwen, J.; Chan, E.; Mattice, M.; Legendre, A.; Kastelein, J.J.P.; Hayden, M.R. Increased Risk of Coronary Artery Disease in Caucasians with Extremely Low HDL Cholesterol Due to Mutations in ABCA1, APOA1, and LCAT. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2012, 1821, 416–424. [Google Scholar] [CrossRef]
- Brunham, L.R.; Hayden, M.R. Human Genetics of HDL: Insight into Particle Metabolism and Function. Prog. Lipid Res. 2015, 58. [Google Scholar] [CrossRef] [Green Version]
- Ikewaki, K.; Rader, D.J.; Sakamoto, T.; Nishiwaki, M.; Wakimoto, N.; Schaefer, J.R.; Ishikawa, T.; Fairwell, T.; Zech, L.A.; Nakamura, H.; et al. Delayed Catabolism of High Density Lipoprotein Apolipoproteins A-I and A-II in Human Cholesteryl Ester Transfer Protein Deficiency. J. Clin. Investig. 1993, 92, 1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, S.; Sharp, D.S.; Grove, J.S.; Bruce, C.; Yano, K.; Curb, J.D.; Tall, A.R. Increased Coronary Heart Disease in Japanese-American Men with Mutation in the Cholesteryl Ester Transfer Protein Gene despite Increased HDL Levels. J. Clin. Investig. 1996, 97, 2917–2923. [Google Scholar] [CrossRef]
- Papp, A.C.; Pinsonneault, J.K.; Wang, D.; Newman, L.C.; Gong, Y.; Johnson, J.A.; Pepine, C.J.; Kumari, M.; Hingorani, A.D.; Talmud, P.J.; et al. Cholesteryl Ester Transfer Protein (CETP) Polymorphisms Affect MRNA Splicing, HDL Levels, and Sex-Dependent Cardiovascular Risk. PLoS ONE 2012, 7, e31930. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S. Unique Features of High-Density Lipoproteins in the Japanese: In Population and in Genetic Factors. Nutrients 2015, 7, 2359–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manichaikul, A.; Wang, X.-Q.; Musani, S.K.; Herrington, D.M.; Post, W.S.; Wilson, J.G.; Rich, S.S.; Rodriguez, A. Association of the Lipoprotein Receptor SCARB1 Common Missense Variant Rs4238001 with Incident Coronary Heart Disease. PLoS ONE 2015, 10, e0125497. [Google Scholar] [CrossRef] [Green Version]
- Zanoni, P.; Khetarpal, S.A.; Larach, D.B.; Hancock-Cerutti, W.F.; Millar, J.S.; Cuchel, M.; DerOhannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; et al. Rare Variant in Scavenger Receptor BI Raises HDL Cholesterol and Increases Risk of Coronary Heart Disease. Science 2016, 351, 1166–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helgadottir, A.; Sulem, P.; Thorgeirsson, G.; Gretarsdottir, S.; Thorleifsson, G.; Jensson, B.Ö.; Arnadottir, G.A.; Olafsson, I.; Eyjolfsson, G.I.; Sigurdardottir, O.; et al. Rare SCARB1 Mutations Associate with High-Density Lipoprotein Cholesterol but Not with Coronary Artery Disease. Eur. Heart J. 2018, 39, 2172–2178. [Google Scholar] [CrossRef] [PubMed]
- Brophy, V.H.; Jarvik, G.P.; Richter, R.J.; Rozek, L.S.; Schellenberg, G.D.; Furlong, C.E. Analysis of Paraoxonase (PON1) L55M Status Requires Both Genotype and Phenotype. Pharmacogenetics 2000, 10, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Garin, M.C.; James, R.W.; Dussoix, P.; Blanché, H.; Passa, P.; Froguel, P.; Ruiz, J. Paraoxonase Polymorphism Met-Leu54 Is Associated with Modified Serum Concentrations of the Enzyme. A Possible Link between the Paraoxonase Gene and Increased Risk of Cardiovascular Disease in Diabetes. J. Clin. Investig. 1997, 99, 62–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-Q.; Ma, N.; Li, X.-G.; Wang, B.; Sun, S.-S.; Gao, F.; Mo, D.-P.; Song, L.-G.; Sun, X.; Liu, L.; et al. Association of PON1, P2Y12 and COX1 with Recurrent Ischemic Events in Patients with Extracranial or Intracranial Stenting. PLoS ONE 2016, 11, e0148891. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Narang, R.; Venkatraman, A.; Das, N. Two- and Three-Locus Haplotypes of the Paraoxonase (PON1) Gene Are Associated with Coronary Artery Disease in Asian Indians. Gene 2012, 506, 242–247. [Google Scholar] [CrossRef]
- Balcerzyk, A.; Zak, I.; Krauze, J. Protective Effect of R Allele of PON1 Gene on the Coronary Artery Disease in the Presence of Specific Genetic Background. Dis. Mark. 2008, 24, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabana, N.; Ashiq, S.; Ijaz, A.; Khalid, F.; ul Saadat, I.; Khan, K.; Sarwar, S.; Shahid, S.U. Genetic Risk Score (GRS) Constructed from Polymorphisms in the PON1, IL-6, ITGB3, and ALDH2 Genes Is Associated with the Risk of Coronary Artery Disease in Pakistani Subjects. Lipids Health Dis. 2018, 17, 224. [Google Scholar] [CrossRef] [Green Version]
- Humbert, R.; Adler, D.A.; Disteche, C.M.; Hassett, C.; Omiecinski, C.J.; Furlong, C.E. The Molecular Basis of the Human Serum Paraoxonase Activity Polymorphism. Nat. Genet. 1993, 3, 73–76. [Google Scholar] [CrossRef]
- Zhao, D.; He, Z.; Qin, X.; Li, L.; Liu, F.; Deng, S. Association of Apolipoprotein M Gene Polymorphisms with Ischemic Stroke in a Han Chinese Population. J. Mol. Neurosci. 2011, 43, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Jiao, G.; Yuan, Z.; Xue, Y.; Yang, C.; Lu, C.; Lü, Z.; Xiao, M. A Prospective Evaluation of Apolipoprotein M Gene T-778C Polymorphism in Relation to Coronary Artery Disease in Han Chinese. Clin. Biochem. 2007, 40, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Luo, G.; Zhang, J.; Mu, Q.; Shi, Y.; Berggren-Söderlund, M.; Nilsson-Ehle, P.; Zhang, X.; Xu, N. Decreased Activities of Apolipoprotein m Promoter Are Associated with the Susceptibility to Coronary Artery Diseases. Int. J. Med. Sci. 2014, 11, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Disease | miRNA Profile | Function | Ref | |
|---|---|---|---|---|
| upregulated | downregulated | |||
| CAD | miR-33a miR-92a * miR-125a miR-146a † miR-486 | miR-33a: inhibits genes involved in cholesterol transport and fatty acid metabolism (decreased cholesterol efflux and fatty acid oxidation). Additionally promotes cardiac fibrosis by targeting matrix metalloproteinase 16. miR-92a: induces endothelial dysfunction and cardiomyocyte apoptosis. miR-125a: inhibits vascular smooth muscle cell proliferation and migration by targeting MAPK1. Endothelial cell metabolic reprogramming (glycolysis) mediates miR-125a-induced vascular hyperbranching. miR-146a: associated with the control of inflammatory processes. miR-486: increases cholesterol accumulation in foam cells. Hypoxia-induced expression. Additionally inhibits cardiomyocyte apoptosis. | [108] | |
| ACS | miR-30c miR-92a * miR-146a † | miR-30c: downregulates the pro-fibrotic connective tissue growth factor, modulating structural changes in the extracellular matrix of the myocardium. miR-92a: pro-inflammatory and angiogenesis-promoting. Highly expressed in endothelial cells. miR-146: see above. | [107] | |
| FH | miR-105 miR-106a miR-223 | miR-105: disrupts vascular integrity (Zonula Ocludens-1 tight junctions). miR-106a: induces cardiac hypertrophy. miR-223: potentially atherogenic and predictive for coronary artery disease. Direct targets: Ras homolog family member B (controls endothelial barrier integrity during inflammation) and ephrin A1 (pro-angiogenic upon hypoxia). | [106] | |
| FH | miR-486 miR-92a * | miR-486: see above. miR-92a: see above. | [109] | |
| Gene | Annotated Mutations | Associated to CV Risk? | Affected Physiological Parameters | Ref |
|---|---|---|---|---|
| APOA-I | 83 | Yes | Mild (heterozygous) to almost complete absence (homozygous or compound heterozygous) of ApoA-I and HDL-C with a predisposition for premature CVD. (Hereditary) amyloidosis due to the accumulation of abnormal N-terminal ApoA-I fragments. | [110,111,112,113,114] |
| ABCA1 | 268 | Yes | Heterozygous loss-of-function: common in people with low HDL-C presenting with a 50% reduction in cholesterol efflux and moderately reduced HDL-C. Homozygous: Tangier Disease counts 100 cases worldwide and shows drastic impairment of cholesterol efflux and hardly any plasma HDL-C and ApoA-I. ABCA1 mutations seem to be dominant: combinations with mutations that increase HDL-C levels, sustain very low HDL-C. Often accompanied by neurologic, ophthalmologic, dermatologic, hematologic, and histiocytic symptoms. | [115,116,117] |
| LCAT | 117 | Expected, but not confirmed due to low case numbers and high heterogeneity. | Mild (fish-eye disease) to severe (familial LCAT deficiency) loss of enzymatic activity resulting in a reduction of ApoA-I and HDL-C plasma levels of up to 80%. | [118,119,120] |
| CETP | 71 | Yes, but extent strongly depends on the respective mutation. | Partial to complete deficiency increases ApoA-I and HDL-C plasma levels. More frequently found in the Japanese population. | [30,121,122,123,124] |
| SCARB1 | 18 | Unclear. Yes for rs4238001 and p.P376L (almost exclusive to Ashkenazi Jews). | Increased HDL-C (impaired hepatic uptake) and foam cell formation (impaired cholesterol efflux). A higher prevalence in the Icelandic population. | [105,125,126,127] |
| PON1 | 22 | Unclear. Yes for Q192R and V109I (ischemic events, CAD) and suggested for L55M (AS) in diabetic patients. | Protection from oxidation diminished by impaired enzymatic activity (Q192R) or reduced concentrations (L55M). | [128,129,130,131,132,133,134] |
| APOM | 5 | Yes for T778C, T1628G, T855C, C724del. | Supposedly down-regulated ApoM expression and elevated total cholesterol levels. Almost exclusively identified in the Han Chinese population. | [135,136,137] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoch, L.; Badimon, L.; Vilahur, G. Unraveling the Complexity of HDL Remodeling: On the Hunt to Restore HDL Quality. Biomedicines 2021, 9, 805. https://doi.org/10.3390/biomedicines9070805
Schoch L, Badimon L, Vilahur G. Unraveling the Complexity of HDL Remodeling: On the Hunt to Restore HDL Quality. Biomedicines. 2021; 9(7):805. https://doi.org/10.3390/biomedicines9070805
Chicago/Turabian StyleSchoch, Leonie, Lina Badimon, and Gemma Vilahur. 2021. "Unraveling the Complexity of HDL Remodeling: On the Hunt to Restore HDL Quality" Biomedicines 9, no. 7: 805. https://doi.org/10.3390/biomedicines9070805