
Neuroinflammation in Alzheimer’s Disease

 and
and 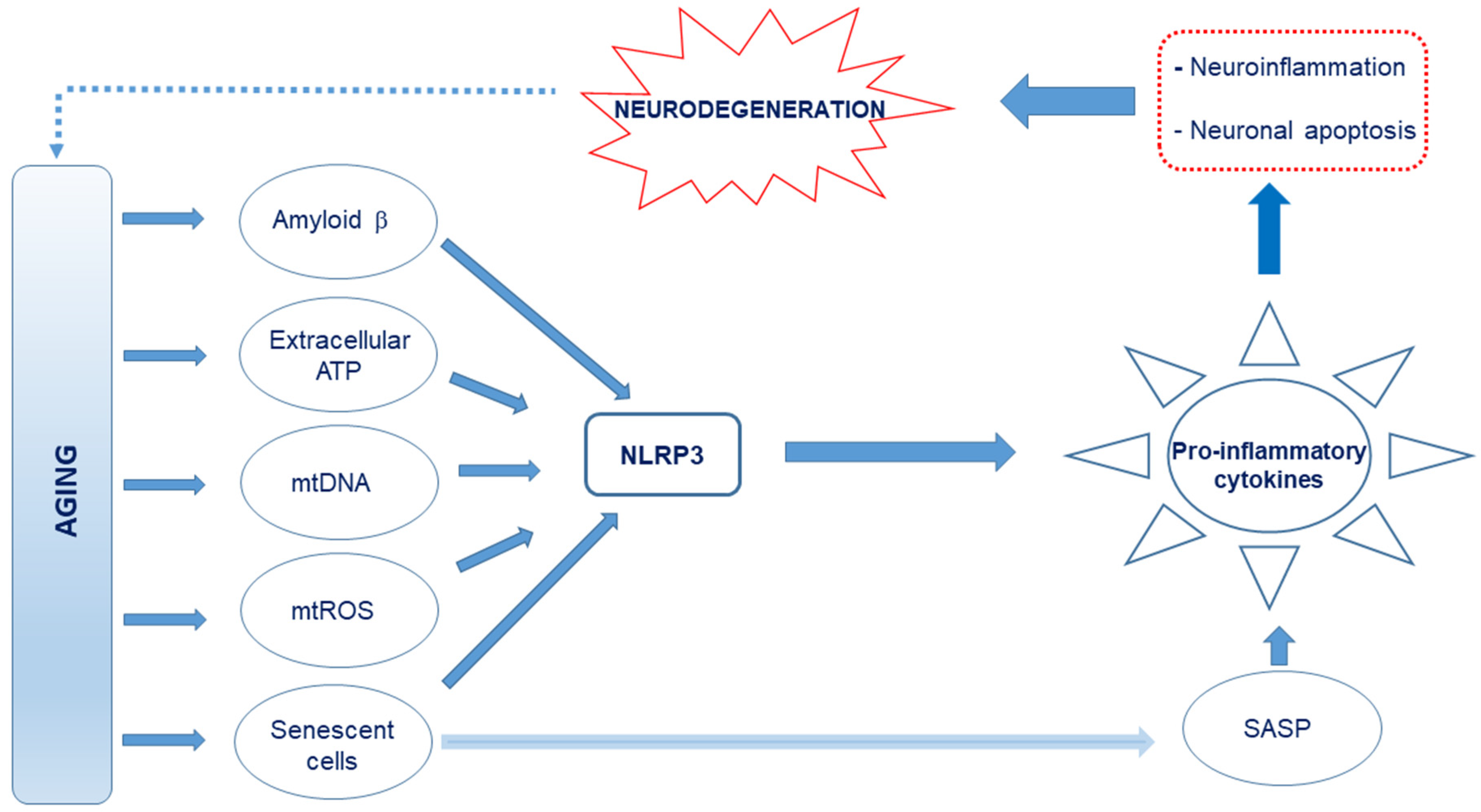{kind=link}
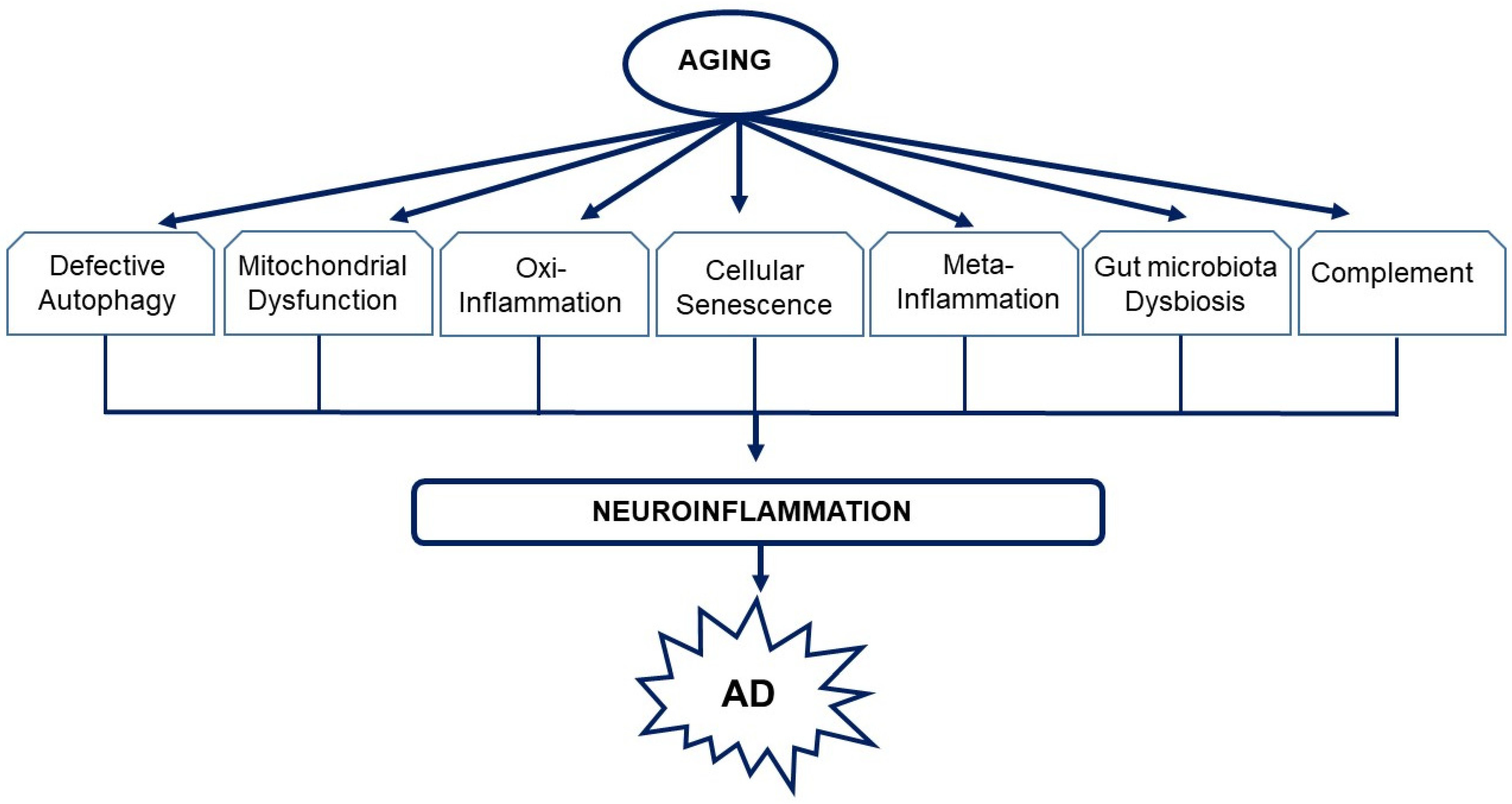{kind=link}
Abstract
:1. Introduction
2. Mediators of Neuroinflammation
2.1. Microglia
2.2. Astrocytes
2.3. Oligodendrocytes
2.4. Myeloid Cells Other Than Microglia
3. Defective Autophagy and Neuroinflammation
4. Mitochondrial Dysfunction and Immunometabolism
5. Oxidative Stress and Neuroinflammation
6. Cellular Senescence and Neuroinflammation
7. Meta-Inflammation (Metaflammation)
8. Exercise and Inflammation
9. Gut Microbiota and Inflammation
10. Complement in the Brain
11. Possible Intervention for Neuroinflammation in AD
11.1. Targeting TNF-α
11.2. Senolytics
11.3. Targeting the Inflammasome
11.4. Targeting Immune Checkpoints
11.5. Targeting CD38
11.6. Targeting CD33
11.7. Targeting IL-12/IL23
11.8. Targeting Th1 Response
11.9. Targeting Microglial P2Y6 Receptor
11.10. Cyclosporine and Tacrolimus
11.11. Targeting p38MAPK
11.12. CNS Targeting Anti-Complement Agents
11.13. Novel Non-Steroidal Anti-Inflammatory Derivatives
11.14. Calorie Restriction (CR)
11.15. Metformin
11.16. Endurance Exercise
11.17. Melatonin
11.18. Resveratrol
11.19. Antioxidants
11.20. Probiotics
11.21. Nutraceuticals
11.22. Essential Vitamins and Minerals
11.23. Flavonoids from Epimedium and Icariin
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| Aβ | Amyloid beta |
| ABCA7 | ATP-binding cassette subfamily A member 7 |
| AD | Alzheimer’s disease |
| Ahr | Aryl hydrocarbon receptor |
| AMPK | AMP-activated kinase |
| APP | Amyloid precursor protein |
| BBB | Blood–brain barrier |
| BIN1 | Bridging integrator 1 |
| BMAA | α-Amino-β-methylaminopropionic acid |
| CCR2 | C-C chemokine receptor type 2 |
| CD | Cluster of differentiation |
| CD2AP | CD2 associated protein |
| CLU | Clusterin |
| CNS | Central nervous system |
| CR | Calorie restriction |
| CR1 | Complement component (3b/4b) receptor 1 |
| CRP | C-reactive protein |
| CSF | Cerebrospinal fluid |
| DAMPs | Damage associated molecular patterns |
| EE | Endurance exercise |
| EOAD | Early-onset Alzheimer’s disease |
| GFAP | Glial fibrillary acidic protein |
| GA | Glatiramer acetate |
| GWAS | Genome-wide association studies |
| HMGB1 | High mobility group box 1 |
| ICR | Inhibitory immune checkpoint receptor |
| IFNγ | Interferon gamma |
| IMiDs | Immunomodulatory imide drugs |
| LOAD | Late-onset Alzheimer’s disease |
| MDA | Malondialdehyde |
| MMP-9 | Matrix metallopeptidase 9 |
| Mo | Monocytes |
| MTOR | Mechanistic target of rapamycin |
| mtROS | Mitochondrial reactive oxygen species |
| MΦ | Macrophages |
| NFκB | Nuclear factor κB |
| NLRP3 | Nod-like receptor 3 |
| NOX | NADPH oxidase |
| OPC | Oligodendrocyte precursor cells |
| PD-1 | Programmed cell death protein-1 |
| PD-L1 | Programmed cell death ligand-1 |
| PICALM | Phosphatidylinositol binding clathrin assembly protein |
| PLD3 | Phospholipase D3 |
| PSEN1 | Presenilin 1 |
| PSEN2 | Presenilin 2 |
| ROI | Reactive oxygen intermediates |
| ROS | Reactive oxygen species |
| RONS | Reactive oxygen and nitrogen species |
| S6RP | S6 ribosomal protein |
| sAPP | Secreted amyloid precursor protein |
| SASP | Senescence-associated secretory phenotype |
| SCFA | Short-chain fatty acids |
| SORL1 | Sortilin-related receptor-1 |
| STAT3 | Signal transducer and activators of transcription 3 |
| T2DM | Type 2 diabetes mellitus |
| TGF-β | Transforming growth factor beta |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor alpha |
| TNFR-I | Tumor necrosis factor receptor I |
| TNFR-II | Tumor necrosis factor receptor II |
| TRAIL | Tumor necrosis factor-related apoptosis inducing ligand |
| Treg | Regulatory T cells |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| VCAM-I | Vascular cell adhesion molecule I |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
| XO | Xanthine oxidase |
References
- Pattabiraman, G.; Palasiewicz, K.; Galvin, J.P.; Ucker, D.S. Aging-associated dysregulation of homeostatic immune response termination (and not initiation). Aging Cell 2017, 16, 585–593. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, R.; Parrillo, L.; Longo, M.; Florese, P.; Desiderio, A.; Zatterale, F.; Miele, C.; Raciti, G.A.; Beguinot, F. Molecular basis of ageing in chronic metabolic diseases. J. Endocrinol. Investig. 2020, 43, 1373–1389. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Li, T.; Huang, Y.; Cai, W.; Chen, X.; Men, X.; Lu, T.; Wu, A.; Lu, Z. Age-related cerebral small vessel disease and inflammaging. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Santoro, A.; Capri, M. The complex relationship between Immunosenescence and Inflammaging: Special issue on the New Biomedical Perspectives. Semin. Immunopathol. 2020, 42, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbé-Tuana, F.; Funchal, G.; Schmitz, C.R.R.; Maurmann, R.M.; Bauer, M.E. The interplay between immunosenescence and age-related diseases. Semin. Immunopathol. 2020, 42, 545–557. [Google Scholar] [CrossRef]
- Conte, M.; Martucci, M.; Chiariello, A.; Franceschi, C.; Salvioli, S. Mitochondria, immunosenescence and inflammaging: A role for mitokines? Semin. Immunopathol. 2020, 42, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.H. Mitochondrial Dysfunction in Aging and Diseases of Aging. Biology 2019, 8, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, M.C.; Grosso, R.A.; Fader, C.M. Hallmarks of Aging: An Autophagic Perspective. Front. Endocrinol. 2019, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling Inflamm-Aging through Mitochondrial Dysfunction: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front. Neurosci. 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Castejon, G. Control of the inflammasome by the ubiquitin system. FEBS J. 2020, 287, 11–26. [Google Scholar] [CrossRef]
- Hegde, A.N.; Smith, S.G.; Duke, L.M.; Pourquoi, A.; Vaz, S. Perturbations of Ubiquitin-Proteasome-Mediated Proteolysis in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 324. [Google Scholar] [CrossRef]
- Ioannidou, A.; Goulielmaki, E.; Garinis, G.A. DNA Damage: From Chronic Inflammation to Age-Related Deterioration. Front. Genet. 2016, 7, 187. [Google Scholar] [CrossRef] [Green Version]
- da Silva, P.F.L.; Schumacher, B. DNA damage responses in ageing. Open Biol. 2019, 9, 190168. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yung, R. Meta-inflammaging at the crossroad of geroscience. Aging Med. 2019, 2, 157–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herradon, G.; Ramos-Alvarez, M.P.; Gramage, E. Connecting Metainflammation and Neuroinflammation Through the PTN-MK-RPTPβ/ζ Axis: Relevance in Therapeutic Development. Front. Pharmacol. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Salvioli, S.; Franceschi, C. Oxidative stress and the ageing endocrine system. Nat. Rev. Endocrinol. 2013, 9, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Shintouo, C.M.; Mets, T.; Beckwee, D.; Bautmans, I.; Ghogomu, S.M.; Souopgui, J.; Leemans, L.; Meriki, H.D.; Njemini, R. Is inflammageing influenced by the microbiota in the aged gut? A systematic review. Exp. Gerontol. 2020, 141, 111079. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Zhao, J.; Wu, L.; Carru, C.; Biagi, E.; Franceschi, C. Microbiomes other than the gut: Inflammaging and age-related diseases. Semin. Immunopathol. 2020, 42, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Fung, E.; Xu, A.; Lan, H.-Y. C-reactive protein and ageing. Clin. Exp. Pharmacol. Physiol. 2017, 44, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Pérez, D.; Sánchez-Flores, M.; Proietti, S.; Bonassi, S.; Costa, S.; Teixeira, J.P.; Fernández-Tajes, J.; Pásaro, E.; Laffon, B.; Valdiglesias, V. Association of inflammatory mediators with frailty status in older adults: Results from a systematic review and meta-analysis. GeroScience 2020, 42, 1451–1473. [Google Scholar] [CrossRef]
- Tchalla, A.E.; Wellenius, G.A.; Travison, T.G.; Gagnon, M.; Iloputaife, I.; Dantoine, T.; Sorond, F.A.; Lipsitz, L.A. Circulating Vascular Cell Adhesion Molecule-1 Is Associated With Cerebral Blood Flow Dysregulation, Mobility Impairment, and Falls in Older Adults. Hypertension 2015, 66, 340–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prochaska, J.H.; Frank, B.; Nagler, M.; Lamparter, H.; Weißer, G.; Schulz, A.; Eggebrecht, L.; Göbel, S.; Arnold, N.; Panova-Noeva, M.; et al. Age-related diagnostic value of D-dimer testing and the role of inflammation in patients with suspected deep vein thrombosis. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Lee, S.-H.; Lee, J.-H.; Lee, H.-Y.; Min, A.K.-J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 4–9. [Google Scholar] [CrossRef]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking Aging to Chronic Disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.-W.; Zhang, X.; Huang, W.-J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, A.L.; Walker, K.A.; Moghekar, A.R.; Pettigrew, C.; Soldan, A.; Albert, M.S.; Walston, J.D. Plasma Markers of Inflammation Linked to Clinical Progression and Decline During Preclinical AD. Front. Aging Neurosci. 2019, 11, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.-Y.; Stachiv, I.; Nikolai, T. Association of Late Life Depression, (Non-) Modifiable Risk and Protective Factors with Dementia and Alzheimer’s Disease: Literature Review on Current Evidences, Preventive Interventions and Possible Future Trends in Prevention and Treatment of Dementia. Int. J. Environ. Res. Public Health 2020, 17, 7475. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J. New approaches to symptomatic treatments for Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 1–13. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Roda, A.R.; Montoliu-Gaya, L.; Serra-Mir, G.; Villegas, S. Both Amyloid-β Peptide and Tau Protein Are Affected by an Anti-Amyloid-β Antibody Fragment in Elderly 3xTg-AD Mice. Int. J. Mol. Sci. 2020, 21, 6630. [Google Scholar] [CrossRef]
- Reitz, C.; Rogaeva, E.; Beecham, G.W. Late-onset vs nonmendelian early-onset Alzheimer disease: A distinction without a difference? Neurol. Genet. 2020, 6, e512. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Cruchaga, C.; Del-Aguila, J.L.; Saef, B.; Black, K.; Fernandez, M.V.; Budde, J.; Ibanez, L.; Deming, Y.; Kapoor, M.; Tosto, G.; et al. Polygenic risk score of sporadic late-onset Alzheimer’s disease reveals a shared architecture with the familial and early-onset forms. Alzheimer’s Dement. 2018, 14, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, R.J., 3rd; Shen, J. Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A An, S.S.; Cai, Y.; Kim, S. Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin. Interv. Aging 2015, 10, 1163–1172. [Google Scholar] [CrossRef] [Green Version]
- Tambini, M.D.; A Norris, K.; D’Adamio, L. Opposite changes in APP processing and human Aβ levels in rats carrying either a protective or a pathogenic APP mutation. eLife 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Vidal, C.; Zhang, L. An Analysis of the Neurological and Molecular Alterations Underlying the Pathogenesis of Alzheimer’s Disease. Cells 2021, 10, 546. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Ryman, D.C.; McDade, E.; Jasielec, M.S.; Buckles, V.D.; Cairns, N.J.; Faga, A.M.; Goate, A.; Marcus, D.S.; Xiong, C.; et al. Neurological manifestations of autosomal dominant familial Alzheimer’s disease: A comparison of the published literature with the dominantly inherited Alzheimer network observational study (DIAN-OBS). Lancet Neurol. 2016, 15, 1317–1325. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, R.; Bras, J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Naj, A.C.; Schellenberg, G.D. Alzheimer’s Disease Genetics Consortium (ADGC). Genomic variants, genes, and pathways of Alzheimer’s disease: An overview. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 5–26. [Google Scholar] [CrossRef] [PubMed]
- Prokopenko, D.; Morgan, S.L.; Mullin, K.; Hofmann, O.; Chapman, B.; Kirchner, R.; Amberkar, S.; Wohlers, I.; Lange, C.; Hide, W.; et al. Whole-genome sequencing reveals new Alzheimer’s disease–associated rare variants in loci related to synaptic function and neuronal development. Alzheimer’s Dement. 2021. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Grozeva, D.; Saad, S.; Menzies, G.E.; Sims, R. Benefits and Challenges of Rare Genetic Variation in Alzheimer’s Disease. Curr. Genet. Med. Rep. 2019, 7, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Lord, J.; Lu, A.J.; Cruchaga, C. Identification of rare variants in Alzheimer’s disease. Front. Genet. 2014, 5, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-Analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Sims, R.; van der Lee, S.J.; Naj, A.C.; Bellenguez, C.; Badarinarayan, N.; Jakobsdottir, J.; Kunkle, B.W.; Boland, A.; Raybould, R.; Bis, J.C.; et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 2017, 49, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.-U.; McCabe, C.; Gjoneska, E.; Sullivan, S.E.; Kaskow, B.J.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 2019, 22, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 2018, 98, 1141–1154. [Google Scholar] [CrossRef] [Green Version]
- Gambhir, I.; Misra, A.; Chakrabarti, S. New genetic players in late-onset Alzheimer’s disease: Findings of genome-wide association studies. Indian J. Med Res. 2018, 148, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.G.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-T.; Zhang, Y. TREM2 regulates innate immunity in Alzheimer’s disease. J. Neuroinflamm. 2018, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Cheng, B.; Li, Y.; Li, X.; Chen, X.; Zhang, Y.-W. TREM2 in Alzheimer’s Disease: Microglial Survival and Energy Metabolism. Front. Aging Neurosci. 2018, 10, 395. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Chen, X.-F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; El Khoury, J. TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 495–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.-X.; Liu, F.; Iqbal, K. Multifactorial Hypothesis and Multi-Targets for Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef]
- De Roeck, A.; Van Broeckhoven, C.; Sleegers, K. The role of ABCA7 in Alzheimer’s disease: Evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019, 138, 201–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, K.; Grundke-Iqbal, I. Alzheimer’s disease, a multifactorial disorder seeking multitherapies. Alzheimer's Dement. 2010, 6, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Sala Frigerio, C.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306. [Google Scholar] [CrossRef] [Green Version]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Shen, Z.; Bao, X.; Wang, R. Clinical PET Imaging of Microglial Activation: Implications for Microglial Therapeutics in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 314. [Google Scholar] [CrossRef]
- Yao, K.; Zu, H.B. Microglial polarization: Novel therapeutic mechanism against Alzheimer’s disease. Inflammopharmacology 2020, 28, 95–110. [Google Scholar] [CrossRef]
- Tondo, G.; Iaccarino, L.; Caminiti, S.P.; Presotto, L.; Santangelo, R.; Iannaccone, S.; Magnani, G.; Perani, D. The combined effects of microglia activation and brain glucose hypometabolism in early-onset Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 1–10. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [PubMed]
- Stamouli, E.C.; Politis, A.M. Pro-inflammatory cytokines in Alzheimer’s disease. Psychiatriki 2016, 27, 264–275. [Google Scholar] [CrossRef]
- Wang, M.-M.; Miao, D.; Cao, X.-P.; Tan, L.; Tan, L. Innate immune activation in Alzheimer’s disease. Ann. Transl. Med. 2018, 6, 177. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Arjona, M.D.M.; Grondona, J.M.; Fernández-Llebrez, P.; López-Ávalos, M.D. Microglial Morphometric Parameters Correlate With the Expression Level of IL-1β, and Allow Identifying Different Activated Morphotypes. Front. Cell Neurosci. 2019, 13, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Azizi, G.; Navabi, S.S.; Al-Shukaili, A.; Seyedzadeh, M.H.; Yazdani, R.; Mirshafiey, A. The Role of Inflammatory Mediators in the Pathogenesis of Alzheimer’s Disease. Sultan Qaboos Univ. Med. J. 2015, 15, e305–e316. [Google Scholar] [CrossRef]
- Chen, X.Q.; Mobley, W.C. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front. Neurosci. 2019, 13, 659. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Rios, M.A.E.; Etoral-Rios, D.; Efranco-Bocanegra, D.; Evilleda-Hernández, J.; Ecampos-Peña, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Businaro, R.; Corsi, M.; Asprino, R.; Di Lorenzo, C.; Laskin, D.; Corbo, R.; Ricci, S.; Pinto, A. Modulation of Inflammation as a Way of Delaying Alzheimer’s Disease Progression: The Diet’s Role. Curr. Alzheimer Res. 2018, 15, 363–380. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Duran-Aniotz, C.; Hetz, C. Glucose Metabolism: A Sweet Relief of Alzheimer’s Disease. Curr. Biol. 2016, 26, R806–R809. [Google Scholar] [CrossRef] [Green Version]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer's Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thal, D.R.; Capetillo-Zarate, E.; Larionov, S.; Staufenbiel, M.; Zurbruegg, S.; Beckmann, N. Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol. Aging 2009, 30, 1936–1948. [Google Scholar] [CrossRef]
- Potter, H.; Granic, A.; Caneus, J. Role of Trisomy 21 Mosaicism in Sporadic and Familial Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 13, 7–17. [Google Scholar] [CrossRef]
- Clare, R.; King, V.G.; Wirenfeldt, M.; Vinters, H.V. Synapse loss in dementias. J. Neurosci. Res. 2010, 88, 2083–2090. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2019, 216, 60–70. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buttgereit, A.; Lelios, I.; Yu, X.; Vrohlings, M.; Krakoski, N.R.; Gautier, E.L.; Nishinakamura, R.; Becher, B.; Greter, M. Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 2016, 17, 1397–1406. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2014, 518, 547–551. [Google Scholar] [CrossRef]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef]
- Sheng, J.; Ruedl, C.; Karjalainen, K. Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity 2015, 43, 382–393. [Google Scholar] [CrossRef] [Green Version]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb+ erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef] [Green Version]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Tay, T.L.; Hagemeyer, N.; Prinz, M. The force awakens: Insights into the origin and formation of microglia. Curr. Opin. Neurobiol. 2016, 39, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.P.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, K.M.; Nelson, L.H. Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mildner, A.; Schmidt, H.; Nitsche, M.; Merkler, D.; Hanisch, U.-K.; Mack, M.; Heikenwalder, M.; Brück, W.; Priller, J.; Prinz, M.; et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 2007, 10, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S.; Powell, N.D.; Godbout, J.P.; Sheridan, J.F. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J. Neurosci. 2013, 33, 13820–13833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sochoka, M.; Diniz, B.S.; Leszek, J. Inflammatory responses in the CNS: Fried or foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejera, D.; Heneka, M.T. Microglia in Alzheimer’s disease: The good, the bad and the ugly. Curr. Alzheimer Res. 2016, 13, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harbor Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W.G.M. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Baroja-Mazo, A.; Martín-Sánchez, F.; Gomez, A.I.; Martínez, C.M.; Amores-Iniesta, J.; Compan, V.; Barberà-Cremades, M.; Yagüe, J.; Ruiz-Ortiz, E.; Antón, J.; et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Koenigsknecht-Talboo, J.; Landreth, G.E. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci. 2005, 25, 8240–8249. [Google Scholar] [CrossRef]
- Sondag, C.M.; Dhawan, G.; Combs, C.K. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J. Neuroinflamm. 2009, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hommet, C.; Mondon, K.; Camus, V.; Ribeiro, M.J.; Beaufils, E.; Arlicot, N.; Corcia, P.; Paccalin, M.; Minier, F.; Gosselin, T.; et al. Neuroinflammation and β Amyloid Deposition in Alzheimer’s Disease: In vivo Quantification with Molecular Imaging. Dement. Geriatr. Cogn. Disord. 2014, 37, 1–18. [Google Scholar] [CrossRef]
- Nagele, R.G.; Wegiel, J.; Venkataraman, V.; Imaki, H.; Wang, K.C.; Wegiel, J. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Mielke, M.L.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 2020, 179, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ries, M.; Sastre, M. Mechanisms of Aβ clearance and degradation by glial cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara, K.; Kim, H.-D.; Jin, J.-J.; Maxwell, J.A.; Li, L.; Fukuchi, K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain J. Neurol. 2006, 129, 3006–3019. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, K.; El Khoury, J. Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer’s disease. Int. J. Alzheimer's Dis. 2012, 2012, 489456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- Thériault, P.; ElAli, A.; Rivest, S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimer's Res. Ther. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, H.L.; Frenkel, D. Immunology and immunotherapy of Alzheimer’s disease. Nat. Rev. Immunol. 2006, 6, 404–416. [Google Scholar] [CrossRef]
- Frackowiak, J.; Wisniewski, H.M.; Wegiel, J.; Merz, G.S.; Iqbal, K.; Wang, K.C. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol. 1992, 84, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef] [PubMed]
- Nagele, R.G.; D’Andrea, M.R.; Lee, H.; Venkataraman, V.; Wang, H.Y. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003, 971, 197–209. [Google Scholar] [CrossRef]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Meda, L.; Baron, P.; Scarlato, G. Glial activation in Alzheimer’s disease: The role of Abeta and its associated proteins. Neurobiol. Aging 2001, 22, 885–893. [Google Scholar] [CrossRef]
- Morgan, D.; Gordon, M.N.; Tan, J.; Wilcock, D.; Rojiani, A.M. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: Implications for Alzheimer therapeutics. J. Neuropathol. Exp. Neurol. 2005, 64, 743–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuppo, E.E.; Arias, H.R. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2005, 37, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.S.; Seshadri, S. Inflammation in the Alzheimer’s disease cascade: Culprit or innocent bystander? Alzheimer's Res. Ther. 2010, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.G.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014, 21, 582–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Jiang, C.; Zou, X.; Zhu, R.; Shi, Y.; Wu, Z.; Zhao, F.; Chen, L. The correlation between accumulation of amyloid beta with enhanced neuroinflammation and cognitive impairment after intraventricular hemorrhage. J. Neurosurg. 2018, 131, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Edison, P.; Archer, H.A.; Gerhard, A.; Hinz, R.; Pavese, N.; Turkheimer, F.E.; Hammers, A.; Tai, Y.F.; Fox, N.; Kennedy, A.; et al. Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol. Dis. 2008, 32, 412–419. [Google Scholar] [CrossRef]
- Okello, A.; Edison, P.; Archer, H.A.; Turkheimer, F.; Kennedy, J.; Bullock, R.; Walker, Z.; Fox, N.; Rossor, M.; Brooks, D.J. Microglial activation and amyloid deposition in mild cognitive impairment: A PET study. Neurology 2009, 72, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; De Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using18F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Okello, A.A.; Brooks, D.J.; Edison, P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain 2015, 138, 3685–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokokura, M.; Mori, N.; Yagi, S.; Yoshikawa, E.; Kikuchi, M.; Yoshihara, Y.; Wakuda, T.; Sugihara, G.; Takebayashi, K.; Suda, S.; et al. In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2010, 38, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Gosztyla, M.L.; Brothers, H.M.; Robinson, S.R. Alzheimer’s Amyloid-β is an Antimicrobial Peptide: A Review of the Evidence. J. Alzheimer's Dis. 2018, 62, 1495–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.-S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Preman, P.; Alfonso-Triguero, M.; Alberdi, E.; Verkhratsky, A.; Arranz, A. Astrocytes in Alzheimer’s Disease: Pathological Significance and Molecular Pathways. Cells 2021, 10, 540. [Google Scholar] [CrossRef] [PubMed]
- Sheeler, C.; Rosa, J.-G.; Ferro, A.; McAdams, B.; Borgenheimer, E.; Cvetanovic, M. Glia in Neurodegeneration: The Housekeeper, the Defender and the Perpetrator. Int. J. Mol. Sci. 2020, 21, 9188. [Google Scholar] [CrossRef]
- Gamage, R.; Wagnon, I.; Rossetti, I.; Childs, R.; Niedermayer, G.; Chesworth, R.; Gyengesi, E. Cholinergic Modulation of Glial Function During Aging and Chronic Neuroinflammation. Front. Cell. Neurosci. 2020, 14, 577912. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, F.; Quintana, F.J. The role of astrocytes in CNS inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Hol, E.M.; Pekny, M. Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr. Opin. Cell Biol. 2015, 32, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, K.K. Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Beach, T.; McGeer, E. Lamina-specific arrangement of astrocytic gliosis and senile plaques in Alzheimer’s disease visual cortex. Brain Res. 1988, 463, 357–361. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Olabarria, M.; Chvatal, A.; Verkhratsky, A.; Rodr, J.J. Astroglia in dementia and Alzheimer’s disease. Cell Death Differ. 2008, 16, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkhratsky, A.; Zorec, R.; Parpura, V. Stratification of astrocytes in healthy and diseased brain. Brain Pathol. 2017, 27, 629–644. [Google Scholar] [CrossRef] [PubMed]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, J.; Quinn, B.; Puissant, V. Disruption of astroglial interlaminar processes in Alzheimer’s disease. Brain Res. Bull. 2002, 58, 235–242. [Google Scholar] [CrossRef]
- Yeh, C.-Y.; Vadhwana, B.; Verkhratsky, A.; Rodriguez, J.J. Early Astrocytic Atrophy in the Entorhinal Cortex of a Triple Transgenic Animal Model of Alzheimer’s Disease. ASN Neuro 2011, 3, e00071. [Google Scholar] [CrossRef] [PubMed]
- Beauquis, J.; Vinuesa, A.; Pomilio, C.; Pavía, P.; Galván, V.; Saravia, F. Neuronal and Glial Alterations, Increased Anxiety, and Cognitive Impairment before Hippocampal Amyloid Deposition in PDAPP Mice, Model of Alzheimer’s Disease. Hippocampus 2014, 24, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Diniz, L.P.; Tortelli, V.; Matias, I.; Morgado, J.; Araujo, A.P.B.; Melo, H.M.; Da Silva, G.S.S.; Alves-Leon, S.V.; De Souza, J.M.; Ferreira, S.T.; et al. Astrocyte Transforming Growth Factor Beta 1 Protects Synapses against Aβ Oligomers in Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 6797–6809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iram, T.; Trudler, D.; Kain, D.; Kanner, S.; Galron, R.; Vassar, R.; Barzilai, A.; Blinder, P.; Fishelson, Z.; Frenkel, D. Astrocytes from old Alzheimer’s disease mice are impaired in Aβ uptake and in neuroprotection. Neurobiol. Dis. 2016, 96, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Polis, B.; Srikanth, K.D.; Elliott, E.; Gil-Henn, H.; Samson, A.O. L-Norvaline Reverses Cognitive Decline and Synaptic Loss in a Murine Model of Alzheimer’s Disease. Neurotherapeutics 2018, 15, 1036–1054. [Google Scholar] [CrossRef] [Green Version]
- Jones, V.C.; Atkinson-Dell, R.; Verkhratsky, A.; Mohamet, L. Aberrant iPSC-Derived Human Astrocytes in Alzheimer’s Disease. Cell Death Dis. 2017, 8, e2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, A.W.; Hu, X.; Yoon, H.; Yan, P.; Xiao, Q.; Wang, Y.; Gil, S.C.; Brown, J.; Wilhelmsson, U.; Restivo, J.L.; et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013, 27, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, E.; Nakano, M.; Kubota, K.; Himuro, N.; Mizoguchi, S.; Chikenji, T.; Otani, M.; Mizue, Y.; Nagaishi, K.; Fujimiya, M. Activated forms of astrocytes with higher GLT-1 expression are associated with cognitive normal subjects with Alzheimer pathology in human brain. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Alford, M.; DeTeresa, R.; Mallory, M.; Hansen, L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1996, 40, 759–766. [Google Scholar] [CrossRef]
- Scimemi, A.; Meabon, J.S.; Woltjer, R.L.; Sullivan, J.M.; Diamond, J.S.; Cook, D.G. Amyloid-β1-42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J. Neurosci. 2013, 33, 5312–5318. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.J.; Gasperini, R.; Foa, L.; Small, D.H. Astrocytes in Alzheimer’s Disease: Emerging Roles in Calcium Dysregulation and Synaptic Plasticity. J. Alzheimer’s Dis. 2010, 22, 699–714. [Google Scholar] [CrossRef]
- Ettle, B.; Schlachetzki, J.C.M.; Winkler, J. Oligodendroglia and myelin in neurodegenerative diseases: More than just bystanders? Mol. Neurobiol. 2016, 53, 3046–3062. [Google Scholar] [CrossRef] [Green Version]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G. Age-related myelin breakdown: A developmental model of cognitive decline and Alzheimer’s disease. Neurobiol. Aging 2004, 25, 5–18. [Google Scholar] [CrossRef]
- Bartzokis, G.; Lu, P.H.; Mintz, J. Quantifying age-related myelin breakdown with MRI: Novel therapeutic targets for preventing cognitive decline and Alzheimer’s disease. J. Alzheimer's Dis. 2004, 6, S53–S59. [Google Scholar] [CrossRef] [PubMed]
- Mitew, S.; Kirkcaldie, M.T.K.; Halliday, G.M.; Shepherd, C.E.; Vickers, J.C.; Dickson, T.C. Focal demyelination in Alzheimer’s disease and transgenic mouse models. Acta Neuropathol. 2010, 119, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.K.; Mastrangelo, M.A.; Ryan, D.A.; Sudol, K.L.; Narrow, W.C.; Bowers, W.J. Early Oligodendrocyte/Myelin Pathology in Alzheimer’s Disease Mice Constitutes a Novel Therapeutic Target. Am. J. Pathol. 2010, 177, 1422–1435. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, H.R.; Ruitenberg, M.J.; McMenamin, P.G. Novel characterization of monocyte-derived cell populations in the meninges and choroid plexus and their rates of replenishment in bone marrow chimeric mice. J. Neuropathol. Exp. Neurol. 2010, 69, 896–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Posse de Chaves, E. Aβ internalization by neurons and glia. Int. J. Alzheimer's Dis. 2011, 2011, 127984. [Google Scholar]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.-P.; Rivest, S. Bone Marrow-Derived Microglia Play a Critical Role in Restricting Senile Plaque Formation in Alzheimer’s Disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Thanopoulou, K.; Fragkouli, A.; Stylianopoulou, F.; Georgopoulos, S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 20816–20821. [Google Scholar] [CrossRef] [Green Version]
- Town, T.; Laouar, Y.; Pittenger, C.; Mori, T.; Szekely, C.A.; Tan, J.; Duman, R.S.; Flavell, R.A. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 2008, 14, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; He, P.; Huang, Y.; Li, Y.-F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef]
- Wang, R.; Wang, G. Autophagy in Mitochondrial Quality Control. Adv. Exp. Med. Biol. 2019, 1206, 421–434. [Google Scholar] [PubMed]
- Harper, J.W.; Ordureau, A.; Heo, J.-M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the Autophagy-Inflammation-Cell Death Axis in Organismal Aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. Mol. Life Sci. 2014, 157, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, S.; Juncker, M.; Kim, C. Regulation of mitophagy by the ubiquitin pathway in neurodegenerative diseases. Exp. Biol. Med. 2018, 243, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Madruga, E.; Maestro, I.; Martínez, A. Mitophagy Modulation, a New Player in the Race against ALS. Int. J. Mol. Sci. 2021, 22, 740. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Dall’Olio, F.; Vanhooren, V.; Chen, C.C.; Slagboom, P.E.; Wuhrer, M.; Franceschi, C. N-glycomic biomarkers of biological aging and longevity: A link with inflammaging. Ageing Res. Rev. 2013, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Wei, X.; Wei, Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell 2016, 7, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanaja, S.K.; Rathinam, V.A.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Mamik, M.K.; Power, C. Inflammasomes in neurological diseases: Emerging pathogenic and therapeutic concepts. Brain 2017, 140, 2273–2285. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Downs, K.P.; Nguyen, H.; Dorfleutner, A.; Stehlik, C. An overview of the non-canonical inflammasome. Mol. Asp. Med. 2020, 76, 100924. [Google Scholar] [CrossRef] [PubMed]
- Horng, T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014, 35, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1{beta} secretion by targeting pro-IL-1{beta} for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Kim, K.H.; Cohen, P. MOTS-c: A novel mitochondrial-derived peptide regulating muscle and fat metabolism. Free. Radic. Biol. Med. 2016, 100, 182–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, D.; Ye, Z.; Jiang, Y.; Xu, C.; Ruan, B.; Yang, Y.; Lei, X.; Xiang, A.; Lu, H.; Zhu, Z.; et al. MOTS-c peptide increases survival and decreases bacterial load in mice infected with MRSA. Mol. Immunol. 2017, 92, 151–160. [Google Scholar] [CrossRef]
- Oh, Y.K.; Bachar, A.R.; Zacharias, D.G.; Kim, S.G.; Wan, J.; Cobb, L.J.; Lerman, L.O.; Cohen, P.; Lerman, A. Humanin preserves endothelial function and prevents atherosclerotic plaque progression in hypercholesterolemic ApoE deficient mice. Atherosclerosis 2011, 219, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Sreekumar, P.G.; Ishikawa, K.; Spee, C.; Mehta, H.H.; Wan, J.; Yen, K.; Cohen, P.; Kannan, R.; Hinton, D.R. The Mitochondrial-Derived Peptide Humanin Protects RPE Cells From Oxidative Stress, Senescence, and Mitochondrial Dysfunction. Investig. Opthalmology Vis. Sci. 2016, 57, 1238–1253. [Google Scholar] [CrossRef]
- Muzumdar, R.H.; Huffman, D.M.; Atzmon, G.; Buettner, C.; Cobb, L.J.; Fishman, S.; Budagov, T.; Cui, L.; Einstein, F.H.; Poduval, A.; et al. Humanin: A Novel Central Regulator of Peripheral Insulin Action. PLoS ONE 2009, 4, e6334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-J.; Yoon, J.-H.; Ryu, J.-H. Mitophagy: A balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016, 49, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., II; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Fairley, L.H.; Wong, J.H.; Barron, A.M. Mitochondrial Regulation of Microglial Immunometabolism in Alzheimer’s Disease. Front. Immunol. 2021, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Koellhoffer, E.C.; McCullough, L.D.; Ritzel, R.M. Old Maids: Aging and Its Impact on Microglia Function. Int. J. Mol. Sci. 2017, 18, 769. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, I.; Jha, S. Mitochondrial Dysfunction and Alzheimer’s Disease: Role of Microglia. Front. Aging Neurosci. 2020, 120, 252. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.-I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e6. [Google Scholar] [CrossRef]
- Afridi, R.; Kim, J.-H.; Rahman, H.; Suk, K. Metabolic Regulation of Glial Phenotypes: Implications in Neuron–Glia Interactions and Neurological Disorders. Front. Cell. Neurosci. 2020, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Lauro, C.; Limatola, C. Metabolic Reprograming of Microglia in the Regulation of the Innate Inflammatory Response. Front. Immunol. 2020, 11, 493. [Google Scholar] [CrossRef] [Green Version]
- Afridi, R.; Lee, W.H.; Suk, K. Microglia Gone Awry: Linking Immunometabolism to Neurodegeneration. Front. Cell Neurosci. 2020, 14, 246. [Google Scholar] [CrossRef]
- Pan, R.Y.; Ma, J.; Kong, X.X.; Wang, X.F.; Li, S.S.; Qi, X.L.; Yan, Y.H.; Cheng, J.; Liu, Q.; Jin, W.; et al. Sodium rutin ameliorates Alzheimer’s disease-like pathology by enhancing microglial amyloid-β clearance. Sci. Adv. 2019, 5, eaau6328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonda, D.J.; Wang, X.; Lee, H.-G.; Smith, M.A.; Perry, G.; Zhu, X. Neuronal failure in Alzheimer’s disease: A view through the oxidative stress looking-glass. Neurosci. Bull. 2014, 30, 243–252. [Google Scholar] [CrossRef]
- Dorey, E.; Chang, N.; Liu, Q.Y.; Yang, Z.; Zhang, W. Apolipoprotein E, amyloid-beta, and neuroinflammation in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.J.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M.Y. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of TNF. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheeseman, K.H.; Slater, T.F. An Introduction to Free-Radical Biochemistry. Br. Med. Bull. 1993, 49, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Marosi, K.; Bori, Z.; Hart, N.; Sárga, L.; Koltai, E.; Radák, Z.; Nyakas, C. Long-term exercise treatment reduces oxidative stress in the hippocampus of aging rats. Neurosci. 2012, 226, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, ume 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Baeeri, M.; Bahadar, H.; Rahimifard, M.; Navaei-Nigjeh, M.; Khorasani, R.; Rezvanfar, M.A.; Gholami, M.; Abdollahi, M. Alpha-Lipoic acid prevents senescence, cell cycle arrest, and inflammatory cues in fibroblasts by inhibiting oxidative stress. Pharm. Res. 2019, 141, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Chausse, B.; Lewen, A.; Poschet, G.; Kann, O. Selective inhibition of mitochondrial respiratory complexes controls the transition of microglia into a neurotoxic phenotype in situ. Brain, Behav. Immun. 2020, 88, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Calvani, R.; Picca, A.; Marini, F.; Biancolillo, A.; Gervasoni, J.; Persichilli, S.; Primiano, A.; Coelho-Junior, H.J.; Bossola, M.; Urbani, A.; et al. A Distinct Pattern of Circulating Amino Acids Characterizes Older Persons with Physical Frailty and Sarcopenia: Results from the BIOSPHERE Study. Nutrients 2018, 10, 1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baierle, M.; Nascimento, S.N.; Moro, A.M.; Brucker, N.; Freitas, F.; Gauer, B.; Durgante, J.; Bordignon, S.; Zibetti, M.; Trentini, C.M.; et al. Relationship between Inflammation and Oxidative Stress and Cognitive Decline in the Institutionalized Elderly. Oxidative Med. Cell. Longev. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, L.; Christofi, F.L.; Wright, V.P.; Bao, S.; Clanton, T.L. Lipoxygenase-dependent superoxide release in skeletal muscle. J. Appl. Physiol. 2004, 97, 661–668. [Google Scholar] [CrossRef] [Green Version]
- De la Fuente, M.; Miquel, J. An update of the oxidation-inflammation theory of aging: The involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 2009, 15, 3003–3026. [Google Scholar] [CrossRef] [PubMed]
- Daulatzai, M.A. Cerebral hypoperfusion and glucose hypometabolism: Key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J. Neurosci. Res. 2017, 95, 943–972. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; D’Adda Di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell. Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2017, 15, 397–408. [Google Scholar] [CrossRef]
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The Senescence-Associated Secretory Phenotype (SASP) in the Challenging Future of Cancer Therapy and Age-Related Diseases. Biology 2020, 9, 485. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neves, J.; Demaria, M.; Campisi, J.; Jasper, H. Of Flies, Mice, and Men: Evolutionarily Conserved Tissue Damage Responses and Aging. Dev. Cell 2015, 32, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borodkina, A.V.; Deryabin, P.I.; Giukova, A.A.; Nikolsky, N.N. ‘Social life’ of senescent sells: What is SASP and why study it? Acta Naturae 2018, 10, 4–14. [Google Scholar] [CrossRef]
- Moreno-Blas, D.; Gorostieta-Salas, E.; Pommer-Alba, A.; Muciño-Hernández, G.; Gerónimo-Olvera, C.; Maciel-Barón, L.A.; Konigsberg, M.; Massieu, L.; Castro-Obregón, S. Cortical neurons develop a senescence-like phenotype promoted by dysfunctional autophagy. Aging 2019, 11, 6175–6198. [Google Scholar] [CrossRef] [PubMed]
- Acklin, S.; Zhang, M.; Du, W.; Zhao, X.; Plotkin, M.; Chang, J.; Campisi, J.; Zhou, D.; Xia, F. Depletion of senescent-like neuronal cells alleviates cisplatin-induced peripheral neuropathy in mice. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Schmeer, C.; Kretz, A.; Wengerodt, D.; Stojiljkovic, M.; Witte, O.W. Dissecting Aging and Senescence—Current Concepts and Open Lessons. Cells 2019, 8, 1446. [Google Scholar] [CrossRef] [Green Version]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Wengerodt, D.; Schmeer, C.; Witte, O.W.; Kretz, A. Amitosenescence and Pseudomitosenescence: Putative New Players in the Aging Process. Cells 2019, 8, 1546. [Google Scholar] [CrossRef] [Green Version]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; Van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nat. Cell Biol. 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; Valentine, J.M.; Sickora, K.R.; Baeuerle, E.; Thompson, C.S.; Shen, Q.; Orr, M.E. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17, e12840. [Google Scholar] [CrossRef] [PubMed]
- Walton, C.C.; Andersen, J.K. Unknown fates of (brain) oxidation or UFO: Close encounters with neuronal senescence. Free Radic. Biol. Med. 2019, 134, 695–701. [Google Scholar] [CrossRef]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Sen, J.M.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef] [Green Version]
- Fafián-Labora, J.A.; O’Loghlen, A. Classical and Nonclassical Intercellular Communication in Senescence and Ageing. Trends Cell Biol. 2020, 30, 628–639. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; An, J.; Zou, M.-H. Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases. Cells 2020, 9, 671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prata, L.G.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Demirci, D.; Dayanc, B.; Mazi, F.; Senturk, S. The Jekyll and Hyde of Cellular Senescence in Cancer. Cells 2021, 10, 208. [Google Scholar] [CrossRef]
- Walton, C.C.; Begelman, D.; Nguyen, W.; Andersen, J.K. Senescence as an Amyloid Cascade: The Amyloid Senescence Hypothesis. Front. Cell. Neurosci. 2020, 14, 14. [Google Scholar] [CrossRef]
- Carreno, G.; Guiho, R.; Martinez-Barbera, J.P. Cell senescence in neuropathology: A focus on neurodegeneration and tumours. Neuropathol. Appl. Neurobiol. 2021, 47, 359–378. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 2012, 7, 45069. [Google Scholar] [CrossRef] [PubMed]
- Pertusa, M.; García-Matas, S.; Rodríguez-Farré, E.; Sanfeliu, C.; Cristòfol, R. Astrocytes aged in vitro show a decreased neuroprotective capacity. J. Neurochem. 2007, 101, 794–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, H.; Chamberlain, C.G.; Weible, M.W.I.; Hughes, S.; Chu, Y.; Chan-Ling, T. Aging-related changes in astrocytes in the rat retina: Imbalance between cell proliferation and cell death reduces astrocyte availability. Aging Cell 2008, 7, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.J.; Wyllie, F.S.; Wynford-Thomas, D.; Kipling, D.; Jones, C.J. A P53-dependent, telomere-independent proliferative life span barrier in human astrocytes consistent with the molecular genetics of glioma development. Cancer Res. 2003, 63, 4854–4861. [Google Scholar] [PubMed]
- Bitto, A.; Sell, C.; Crowe, E.; Lorenzini, A.; Malaguti, M.; Hrelia, S.; Torres, C. Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 2010, 316, 2961–2968. [Google Scholar] [CrossRef]
- Oddo, S.C.A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Vanzulli, I.; Papanikolaou, M.; De-La-Rocha, I.C.; Pieropan, F.; Rivera, A.D.; Gomez-Nicola, D.; Verkhratsky, A.; Rodríguez, J.J.; Butt, A.M. Disruption of oligodendrocyte progenitor cells is an early sign of pathology in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2020, 94, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Luterman, J.D.; Haroutunian, V.; Yemul, S.; Ho, L.; Purohit, D.; Aisen, P.S.; Mohs, R.; Pasinetti, G.M. Cytokine gene expression as a function of the clinical progression of Alzheimer disease dementia. Arch Neurol. 2000, 57, 1153–1160. [Google Scholar] [CrossRef] [Green Version]
- Gruol, D.L. IL-6 regulation of synaptic function in the CNS. Neuropharmacology 2015, 96, 42–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 2013, 210, 2057–2069. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, A.; Burton, D.G.A.; Moshayev, Z.; Vadai, E.; Wensveen, F.; Ben-Dor, S.; Golani, O.; Polic, B.; Krizhanovsky, V. NKG2D ligands mediate immunosurveillance of senescent cells. Aging 2016, 8, 328–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, B.I.; Devine, O.P.; Vukmanovic-Stejic, M.; Chambers, E.S.; Subramanian, P.; Patel, N.; Virasami, A.; Sebire, N.J.; Kinsler, V.; Valdovinos, A.; et al. Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell inhibition. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. XProgrammed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galea, I.; Bechmann, I.; Perry, V.H. What is immune privilege (not)? Trends Immunol. 2007, 28, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Korin, B.; Ben-Shaanan, T.L.; Schiller, M.; Dubovik, T.; Azulay-Debby, H.; Boshnak, N.T.; Koren, T.; Rolls, A. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 2017, 20, 1300–1309. [Google Scholar] [CrossRef]
- Benakis, C.; Llovera, G.; Liesz, A. The meningeal and choroidal infiltration routes for leukocytes in stroke. Ther. Adv. Neurol. Disord. 2018, 11, 11. [Google Scholar] [CrossRef]
- Ardura-Fabregat, A.; Boddeke, E.W.G.M.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzériat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T.; et al. Targeting Neuroinflammation to Treat Alzheimer’s Disease. CNS Drugs 2017, 31, 1057–1082. [Google Scholar] [CrossRef] [Green Version]
- Gorlé, N.; Van Cauwenberghe, C.; Libert, C.; Vandenbroucke, R.E. The effect of aging on brain barriers and the consequences for Alzheimer’s disease development. Mamm. Genome 2016, 27, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef] [PubMed]
- van de Haar, H.J.; Burgmans, S.; Jansen, J.F.; van Osch, M.J.; van Buchem, M.A.; Muller, M.; Hofman, P.A.; Verhey, F.R.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef]
- McManus, R.M.; Mills, K.H.; Lynch, M.A. T cells—protective or pathogenic in Alzheimer’s disease? J. Neuroimmune Pharmacol. 2015, 10, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.T.; Xu, W.; Tan, C.C.; Andrieu, S.; Suckling, J.; Evangelou, E.; Pan, A.; Zhang, C.; Jia, J.; Feng, L.; et al. Evidence-based prevention of Alzheimer’s disease: Systematic review and meta-analysis of 243 observational prospective studies and 153 randomised controlled trials. J. Neurol. Neurosurg. Psychiatry 2020, 111, 1201–1209. [Google Scholar] [CrossRef]
- Kivipelto, M.; Mangialasche, F.; Ngandu, T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 653–666. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Giau, V.V. Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Ji, Y.; Kersten, S.; Qi, L. Mechanisms of Inflammatory Responses in Obese Adipose Tissue. Annu. Rev. Nutr. 2012, 32, 261–286. [Google Scholar] [CrossRef] [Green Version]
- Mraz, M.; Haluzik, M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J. Endocrinol. 2014, 222, R113–R127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenk, S.; Saberi, M.; Olefsky, J.M. Insulin sensitivity: Modulation by nutrients and inflammation. J. Clin. Investig. 2008, 118, 2992–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.T.D.; Le, T.T.; Vo, V.G. Role of Insulin Resistance in the Alzheimer’s Disease Progression. Neurochem. Res. 2020, 45, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Bahniwal, M.; Little, J.P.; Klegeris, A. High Glucose Enhances Neurotoxicity and Inflammatory Cytokine Secretion by Stimulated Human Astrocytes. Curr. Alzheimer Res. 2017, 14, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Anjum, I.; Fayyaz, M.; Wajid, A.; Sohail, W.; Ali, A. Does Obesity Increase the Risk of Dementia: A Literature Review. Cureus 2018, 10, e2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Dorantes, M.T.; Díaz-López, Y.E.; Gutiérrez-Aguilar, R. Environment and Gene Association With Obesity and Their Impact on Neurodegenerative and Neurodevelopmental Diseases. Front. Neurosci. 2020, 28, 863. [Google Scholar] [CrossRef] [PubMed]
- Batatinha, H.A.; Biondo, L.A.; Lira, F.S.; Castell, L.M.; Rosa-Neto, J.C. Nutrients, immune system, and exercise: Where will it take us? Nutrition 2019, 61, 151–156. [Google Scholar] [CrossRef]
- Nieman, D.C.; Lila, M.A.; Gillitt, N.D. Immunometabolism: A Multi-Omics Approach to Interpreting the Influence of Exercise and Diet on the Immune System. Annu. Rev. Food Sci. Technol. 2019, 10, 341–363. [Google Scholar] [CrossRef]
- Simpson, R.J.; Lowder, T.W.; Spielmann, G.; Bigley, A.B.; LaVoy, E.C.; Kunz, H. Exercise and the aging immune system. Ageing Res. Rev. 2012, 11, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, K.; Rohde, T.; Zacho, M.; Asp, S.; Pedersen, B.K. Evidence that interleukin-6 is produced in human skeletal muscle during prolonged running. J. Physiol. 1998, 508, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, H.; Yin, Y.-L.; Guo, W.-Z.; Ma, Y.-Q.; Wang, Y.-B.; Shu, C.; Dong, L.-Q. Role of interleukin-6 to differentiate sepsis from non-infectious systemic inflammatory response syndrome. Cytokine 2016, 88, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.A.; Hiscock, N.; Sacchetti, M.; Fischer, C.P.; Pedersen, B.K. Interleukin-6 Is a Novel Factor Mediating Glucose Homeostasis During Skeletal Muscle Contraction. Diabetes 2004, 53, 1643–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, E.W.; Carey, A.L.; Sacchetti, M.; Steinberg, G.R.; Macaulay, S.L.; Febbraio, M.A.; Pedersen, B.K. Acute IL-6 treatment increases fatty acid turnover in elderly humans in vivo and in tissue culture in vitro. Am. J. Physiol. Metab. 2005, 288, E155–E162. [Google Scholar] [CrossRef] [PubMed]
- Neto, J.C.; Silveira, L.S. Endurance Exercise Mitigates Immunometabolic Adipose Tissue Disturbances in Cancer and Obesity. Int. J. Mol. Sci. 2020, 21, 9745. [Google Scholar] [CrossRef]
- Kwilasz, A.; Grace, P.; Serbedzija, P.; Maier, S.; Watkins, L. The therapeutic potential of interleukin-10 in neuroimmune diseases. Neuropharmacology 2015, 96, 55–69. [Google Scholar] [CrossRef] [Green Version]
- Lobo-Silva, D.; Carriche, G.M.; Gil Castro, A.; Roque, S.; Saraiva, M. Balancing the immune response in the brain: IL-10 and its regulation. J. Neuroinflamm. 2016, 13, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Littlefield, A.M.; Setti, S.E.; Priester, C.; Kohman, R.A. Voluntary exercise attenuates LPS-induced reductions in neurogenesis and increases microglia expression of a proneurogenic phenotype in aged mice. J. Neuroinflamm. 2015, 12, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rêgo, M.L.; Cabral, D.A.; Costa, E.C.; Fontes, E.B. Physical Exercise for Individuals with Hypertension: It Is Time to Emphasize its Benefits on the Brain and Cognition. Clin. Med. Insights Cardiol. 2019, 13, 1179546819839411. [Google Scholar] [CrossRef]
- Di Liegro, C.M.; Schiera, G.; Proia, P.; Di Liegro, I. Physical Activity and Brain Health. Genes 2019, 10, 720. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, P.W.; Jeffery, I.B. Gut microbiota and aging. Science 2015, 350, 1214–1215. [Google Scholar] [CrossRef]
- Vemuri, R.; Gundamaraju, R.; Shastri, M.D.; Shukla, S.D.; Kalpurath, K.; Ball, M.; Tristram, S.; Shankar, E.M.; Ahuja, K.; Eri, R. Gut Microbial Changes, Interactions, and Their Implications on Human Lifecycle: An Ageing Perspective. BioMed Res. Int. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Radjabzadeh, D.; Boer, C.G.; Beth, S.A.; Van Der Wal, P.; Jong, J.C.K.-D.; Jansen, M.A.E.; Konstantinov, S.R.; Peppelenbosch, M.P.; Hays, J.P.; Jaddoe, V.W.V.; et al. Diversity, compositional and functional differences between gut microbiota of children and adults. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.B.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimer's Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bostanciklioğlu, M. The role of gut microbiota in pathogenesis of Alzheimer’s disease. J. Appl. Microbiol. 2019, 127, 954–967. [Google Scholar] [CrossRef] [PubMed]
- Talwar, P.; Kushwaha, S.; Gupta, R.; Agarwal, R. Systemic Immune Dyshomeostasis Model and Pathways in Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaz, B.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Schneeberger, M.; Matheis, F.; Wang, P.; Kerner, Z.; Ilanges, A.; Pellegrino, K.; Del Mármol, J.; Castro, T.B.R.; Furuichi, M.; et al. Microbiota modulate sympathetic neurons via a gut–brain circuit. Nat. Cell Biol. 2020, 583, 441–446. [Google Scholar] [CrossRef]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef] [PubMed]
- Rea, K.; Dinan, T.G.; Cryan, J.F. The microbiome: A key regulator of stress and neuroinflammation. Neurobiol. Stress 2016, 4, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Buchanan, J.B.; Sparkman, N.L.; Godbout, J.P.; Freund, G.G.; Johnson, R.W. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav. Immun. 2008, 22, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McManus, R.M.; Heneka, M.T. Role of neuroinflammation in neurodegeneration: New insights. Alzheimer's Res. Ther. 2017, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Carpanini, S.M.; Torvell, M.; Morgan, B.P. Therapeutic Inhibition of the Complement System in Diseases of the Central Nervous System. Front. Immunol. 2019, 10, 362. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P. Complement in the pathogenesis of Alzheimer’s disease. Semin. Immunopathol. 2018, 40, 113–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenner, A.J.; Stevens, B.; Woodruff, T.M. New tricks for an ancient system: Physiological and pathological roles of complement in the CNS. Mol. Immunol. 2018, 102, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.M.; et al. Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep. 2019, 28, e6-2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Takeda, S.; Sato, N.; Morishita, R. Systemic inflammation, blood-brain barrier vulnerability and cognitive/non-cognitive symptoms in Alzheimer disease: Relevance to pathogenesis and therapy. Front. Aging Neurosci. 2014, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, L.B.; Dohgu, S.; Sultana, R.; Lynch, J.L.; Owen, J.B.; Erickson, M.A.; Shah, G.N.; Price, T.O.; Fleegal-Demotta, M.A.; Butterfield, D.A.; et al. Lipopolysaccharide alters the blood-brain barrier transport of amyloid beta protein: A mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav. Immun. 2009, 23, 507–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Propson, N.E.; Roy, E.R.; Litvinchuk, A.; Köhl, J.; Zheng, H. Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J. Clin. Investig. 2021, 131, 131. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, K.; Ahmad, S.; Kindelin, A.; Ducruet, A.F. Complement C3a receptor-mediated vascular dysfunction: A complex interplay between aging and neurodegeneration. J. Clin. Investig. 2021, 131, e144348. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seol, Y.; Ki, S.; Ryu, H.L.; Chung, S.; Lee, J.; Ryu, H. How Microglia Manages Non-cell Autonomous Vicious Cycling of Aβ Toxicity in the Pathogenesis of AD. Front. Mol. Neurosci. 2020, 13, 13. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Hyman, B.T.; Spires-Jones, T.L. Beyond the neuron–cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef]
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steeland, S.; Vandenbroucke, R.E.J. Choroid plexus tumor necrosis factor receptor 1: A new neuroinflammatory piece of the complex Alzheimer’s disease puzzle. Neural Regen Res. 2019, 14, 1144–1147. [Google Scholar] [PubMed]
- Belarbi, K.; Jopson, T.; Tweedie, D.; Arellano, C.; Luo, W.; Greig, N.H.; Rosi, S. TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J. Neuroinflamm. 2012, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Olmos, G.; Lladó, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Yee, K.L.; Sumbria, R.K. Tumor necrosis factor α Inhibition for Alzheimer’s Disease. J. Cent. Nerv. Syst. Dis. 2017, 9, 1179573517709278. [Google Scholar] [CrossRef] [PubMed]
- Paouri, E.; Tzara, O.; Kartalou, G.-I.; Zenelak, S.; Georgopoulos, S. Peripheral Tumor Necrosis Factor-Alpha (TNF-α) Modulates Amyloid Pathology by Regulating Blood-Derived Immune Cells and Glial Response in the Brain of AD/TNF Transgenic Mice. J. Neurosci. 2017, 37, 5155–5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paouri, E.; Tzara, O.; Zenelak, S.; Georgopoulos, S. Genetic Deletion of Tumor Necrosis Factor-α Attenuates Amyloid-β Production and Decreases Amyloid Plaque Formation and Glial Response in the 5XFAD Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Gorlé, N.; VandenDriessche, C.; Balusu, S.; Brkic, M.; Van Cauwenberghe, C.; Van Imschoot, G.; Van Wonterghem, E.; De Rycke, R.; Kremer, A.; et al. Counteracting the effects of TNF receptor-1 has therapeutic potential in Alzheimer’s disease. EMBO Mol. Med. 2018, 10, e8300. [Google Scholar] [CrossRef] [PubMed]
- Lapadula, G.; Marchesoni, A.; Armuzzi, A.; Blandizzi, C.; Caporali, R.; Chimenti, S.; Cimaz, R.; Cimino, L.; Gionchetti, P.; Girolomoni, G.; et al. Adalimumab in the Treatment of Immune-Mediated Diseases. Int. J. Immunopathol. Pharmacol. 2014, 27, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Decourt, B.; Drumm-Gurnee, D.; Wilson, J.; Jacobson, S.; Belden, C.; Sirrel, S.; Ahmadi, M.; Shill, H.; Powell, J.; Walker, A.; et al. Poor Safety and Tolerability Hamper Reaching a Potentially Therapeutic Dose in the Use of Thalidomide for Alzheimer’s disease: Results from a Double-Blind, Placebo-Controlled Trial. Curr. Alzheimer Res. 2017, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Lee, S.-Y.; Shon, J.; Kim, K.; Lee, H.J.; Kim, K.A.; Lee, B.-Y.; Oh, S.-H.; Kim, N.K.; Kim, O.J. Adalimumab improves cognitive impairment, exerts neuroprotective effects and attenuates neuroinflammation in an Aβ1-40-injected mouse model of Alzheimer’s disease. Cytotherapy 2019, 21, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Rivest, S. Alzheimer’s disease: Microglia targets and their modulation to promote amyloid phagocytosis and mitigate neuroinflammation. Expert Opin. Ther. Targets 2020, 24, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xu, R.; Kaelber, D.C.; Gurney, M.E. Tumor Necrosis Factor (TNF) blocking agents are associated with lower risk for Alzheimer’s disease in patients with rheumatoid arthritis and psoriasis. PLoS ONE 2020, 15, e0229819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steed, P.M.; Tansey, M.G.; Zalevsky, J.; Zhukovsky, E.A.; DesJarlais, J.R.; Szymkowski, D.E.; Abbott, C.; Carmichael, D.; Chan, C.; Cherry, L.; et al. Inactivation of TNF Signaling by Rationally Designed Dominant-Negative TNF Variants. Science 2003, 301, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, C.; Tse, Y.C.; Nguyen, H.-B.; Krantic, S.; Breitner, J.C.; Quirion, R.; Wong, T.P. Inhibiting tumor necrosis factor-α before amyloidosis prevents synaptic deficits in an Alzheimer’s disease model. Neurobiol. Aging 2016, 47, 41–49. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, K.P.; Sompol, P.; Kannarkat, G.T.; Chang, J.; Sniffen, L.; Wildner, M.E.; Norris, C.M.; Tansey, M.G. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 2017, 102, 81–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAlpine, F.E.; Lee, J.K.; Harms, A.S.; Ruhn, K.A.; Blurton-Jones, M.; Hong, J.; Das, P.; Golde, T.E.; LaFerla, F.M.; Oddo, S.; et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 2009, 34, 163–177. [Google Scholar] [CrossRef] [Green Version]
- Sama, D.M.; Mohmmad Abdul, H.; Furman, J.L.; Artiushin, I.A.; Szymkowski, D.E.; Scheff, S.W.; Norris, C.M. Inhibition of soluble tumor necrosis factor ameliorates synaptic alterations and Ca2+ dysregulation in aged rats. PLoS ONE 2012, 7, e38170. [Google Scholar] [CrossRef] [Green Version]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.J.; Tweedie, D.; Scerba, M.T.; Greig, N.H. Neuroinflammation as a Factor of Neurodegenerative Disease: Thalidomide Analogs as Treatments. Front. Cell Dev. Biol. 2019, 7, 313. [Google Scholar] [CrossRef]
- He, P.; Cheng, X.; Staufenbiel, M.; Li, R.; Shen, Y. Long-Term Treatment of Thalidomide Ameliorates Amyloid-Like Pathology through Inhibition of β-Secretase in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2013, 8, e55091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decourt, B.; Wilson, J.; Ritter, A.; Dardis, C.; DiFilippo, F.P.; Zhuang, X.; Cordes, D.; Lee, G.; Fulkerson, N.D.; Rose, T.S.; et al. MCLENA-1: A Phase II Clinical Trial for the Assessment of Safety, Tolerability, and Efficacy of Lenalidomide in Patients with Mild Cognitive Impairment Due to Alzheimer’s Disease. Open Access J. Clin. Trials 2020, ume 12, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tweedie, D.; Ferguson, R.A.; Fishman, K.; Frankola, K.A.; Van Praag, H.; Holloway, H.W.; Luo, W.; Li, Y.; Caracciolo, L.; Russo, I.; et al. Tumor necrosis factor-α synthesis inhibitor 3,6’-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.T.; Lecca, D.; Yang, L.Y.; Luo, W.; Scerba, M.T.; Tweedie, D.; Huang, P.S.; Jung, Y.J.; Kim, D.S.; Yang, C.H.; et al. 3,6’-dithiopomalidomide reduces neural loss, inflammation, behavioral deficits in brain injury and microglial activation. Elife 2020, 9, e54726. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Caracciolo, L.; Tweedie, D.; Choi, S.-H.; Greig, N.H.; Barlati, S.; Bosetti, F. 3,6′-Dithiothalidomide, a new TNF-α synthesis inhibitor, attenuates the effect of Aβ1-42 intracerebroventricular injection on hippocampal neurogenesis and memory deficit. J. Neurochem. 2012, 122, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Gabbita, S.P.; Srivastava, M.K.; Eslami, P.; Johnson, M.F.; Kobritz, N.K.; Tweedie, D.; Greig, N.H.; Zemlan, F.P.; Sharma, S.P.; Harris-White, M.E. Early intervention with a small molecule inhibitor for tumor necrosis factor-α prevents cognitive deficits in a triple transgenic mouse model of Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 236. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Cellular senescence: A translational perspective. eBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef] [Green Version]
- Farr, J.N.; Fraser, D.G.; Wang, H.; Jaehn, K.; Ogrodnik, M.B.; Weivoda, M.M.; Drake, M.T.; Tchkonia, T.; Lebrasseur, N.K.; Kirkland, J.L.; et al. Identification of Senescent Cells in the Bone Microenvironment. J. Bone Miner. Res. 2016, 31, 1920–1929. [Google Scholar] [CrossRef]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 5, 4172. [Google Scholar] [CrossRef] [PubMed]
- Sabogal-Guaqueta, A.M.; Munoz-Manco, J.I.; Ramirez-Pineda, J.R.; Lamprea-Rodriguez, M.; Osorio, E.; Cardona-Gomez, G.P. The flavonoid quercetin ameliorates Alzheimer’s disease pathology and protects cognitive and emotional function in aged triple transgenic Alzheimer’s disease model mice. Neuropharmacology 2015, 93, 134–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef] [PubMed]
- Wissler Gerdes, E.O.; Zhu, Y.; Weigand, B.M.; Tripathi, U.; Burns, T.C.; Tchkonia, T.; Kirkland, J.L. Cellular senescence in aging and age-related diseases: Implications for neurodegenerative diseases. Int. Rev. Neurobiol. 2020, 155, 203–234. [Google Scholar] [PubMed]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbarino, V.R.; Tran, S.; E Glassman, J.; Kirkland, J.L.; Musi, N.; Seshadri, S.; E Orr, M. A head-to-head comparison between senolytic therapies, dasatinib plus quercetin and fisetin, indicates sex- and genotype-specific differences in translationally relevant outcomes. Alzheimer’s Dement. 2020, 16. [Google Scholar] [CrossRef]
- Jiang, H.; Gong, T.; Zhou, R. The strategies of targeting the NLRP3 inflammasome to treat inflammatory diseases. Adv. Immunol. 2020, 145, 55–93. [Google Scholar] [CrossRef]
- Chauhan, D.; Walle, L.V.; Lamkanfi, M. Therapeutic modulation of inflammasome pathways. Immunol. Rev. 2020, 297, 123–138. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Goldberg, E.L.; Dixit, V.D. Drivers of age-related inflammation and strategies for healthspan extension. Immunol Rev. 2015, 265, 63–74. [Google Scholar] [CrossRef]
- El-Sharkawy, L.Y.; Brough, D.; Freeman, S. Inhibiting the NLRP3 Inflammasome. Molecules 2020, 25, 5533. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Li, R.; Lyu, J.; Li, X.; Wang, W.; Wang, Z.; Sheng, H.; Zhang, W.; Karhausen, J.; Yang, W. MCC950, a selective NLPR3 inflammasome inhibitor, improves neurologic function and survival after cardiac arrest and resuscitation. J. Neuroinflamm. 2020, 17, 256. [Google Scholar] [CrossRef]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.; Cooper, M.A.; O’Neill, L.A.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Riella, L.V.; Paterson, A.M.; Sharpe, A.H.; Chandraker, A. Role of the PD-1 Pathway in the Immune Response. Arab. Archaeol. Epigr. 2012, 12, 2575–2587. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ji, R.-R. Anti-PD-1 treatment as a neurotherapy to enhance neuronal excitability, synaptic plasticity and memory. bioRxiv 2019, 2019. [Google Scholar] [CrossRef]
- Laumet, G.; Ma, J.; Robison, A.J.; Kumari, S.; Heijnen, C.J.; Kavelaars, A. T Cells as an Emerging Target for Chronic Pain Therapy. Front. Mol. Neurosci. 2019, 12, 216. [Google Scholar] [CrossRef] [Green Version]
- Chamoto, K.; Al-Habsi, M.; Honjo, T. Role of PD-1 in Immunity and Diseases. Curr. Top. Microbiol. Immunol. 2017, 410, 75–97. [Google Scholar] [CrossRef] [PubMed]
- Curdy, N.; Lanvin, O.; Laurent, C.; Fournié, J.-J.; Franchini, D.-M. Regulatory Mechanisms of Inhibitory Immune Checkpoint Receptors Expression. Trends Cell Biol. 2019, 29, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Baruch, K.; Rosenzweig, N.; Kertser, A.; Deczkowska, A.; Sharif, A.M.; Spinrad, A.; Tsitsou-Kampeli, A.; Sarel, A.; Cahalon, L.; Schwartz, M. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer’s disease pathology. Nat. Commun. 2015, 6, 7967. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, M.; González-Cao, M.; Viteri, S.; Karachaliou, N.; Altavilla, G.; Rosell, R. Programmed cell death protein-1/programmed cell death ligand-1 pathway inhibition and predictive biomarkers: Understanding transforming growth factor-beta role. Transl. Lung Cancer Res. 2015, 4, 728–742. [Google Scholar] [PubMed]
- Schwartz, M.; Arad, M.; Ben-Yehuda, H. Potential immunotherapy for Alzheimer disease and age-related dementia. Dialog. Clin. Neurosci 2019, 21, 21–25. [Google Scholar]
- Munafò, A.; Burgaletto, C.; Di Benedetto, G.; Di Mauro, M.; Di Mauro, R.; Bernardini, R.; Cantarella, G. Repositioning of Immunomodulators: A Ray of Hope for Alzheimer’s Disease? Front. Neurosci. 2020, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Baruch, K.; Deczkowska, A.; Rosenzweig, N.; Tsitsou-Kampeli, A.; Sharif, A.M.; Matcovitch-Natan, O.; Kertser, A.; David, E.; Amit, I.; Schwartz, M. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat. Med. 2016, 22, 135–137. [Google Scholar] [CrossRef]
- Rogers, N.K.; Romero, C.; Sanmartín, C.D.; Ponce, D.P.; Salech, F.; López, M.N.; Gleisner, A.; Tempio, F.; Behrens, M.I. Inverse Relationship Between Alzheimer’s Disease and Cancer: How Immune Checkpoints Might Explain the Mechanisms Underlying Age-Related Diseases. J. Alzheimer’s Dis. 2020, 73, 443–454. [Google Scholar] [CrossRef]
- Rosenzweig, N.; Dvir-Szternfeld, R.; Tsitsou-Kampeli, A.; Keren-Shaul, H.; Ben-Yehuda, H.; Weill-Raynal, P.; Cahalon, L.; Kertser, A.; Baruch, K.; Amit, I.; et al. PD-1/PD-L1 checkpoint blockade harnesses monocyte-derived macrophages to combat cognitive impairment in a tauopathy mouse model. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Latta-Mahieu, M.; Elmer, B.; Bretteville, A.; Wang, Y.; Lopez-Grancha, M.; Goniot, P.; Moindrot, N.; Ferrari, P.; Blanc, V.; Schussler, N.; et al. Systemic immune-checkpoint blockade with anti-PD1 antibodies does not alter cerebral amyloid-β burden in several amyloid transgenic mouse models. Glia 2017, 66, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Obst, J.; Mancuso, R.; Simon, E.; Gomez-Nicola, D. PD-1 deficiency is not sufficient to induce myeloid mobilization to the brain or alter the inflammatory profile during chronic neurodegeneration. Brain Behav. Immun. 2018, 73, 708–716. [Google Scholar] [CrossRef]
- Li, S.; Hayden, E.Y.; Garcia, V.J.; Fuchs, D.-T.; Sheyn, J.; Daley, D.A.; Rentsendorj, A.; Torbati, T.; Black, K.L.; Rutishauser, U.; et al. Activated bone marrow-derived macrophages eradicate Alzheimer’s-related Aβ42 Oligomers and protect synapses. Front. Immunol. 2020, 11, 49. [Google Scholar] [CrossRef]
- van de Donk, N.W.C.J. Immunomodulatory effects of CD38-targeting antibodies. Immunol. Lett. 2018, 199, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, S.; Privat, A.-L.; Bressac, L.; Toulorge, D. CD38 in Neurodegeneration and Neuroinflammation. Cells 2020, 9, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blacher, E.; Dadali, T.; Bespalko, A.; Msc, V.J.H.; Grimm, M.O.W.; Hartmann, T.; Lund, F.E.; Stein, R.; Levy, A. Alzheimer’s disease pathology is attenuated in a CD38-deficient mouse model. Ann. Neurol. 2015, 78, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L. CD33 in Alzheimer’s Disease—Biology, Pathogenesis, and Therapeutics: A Mini-Review. Gerontology 2019, 65, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Simpson, J.F.; Parikh, I.; Wilfred, B.R.; Fardo, D.W.; Nelson, P.T.; Estus, S. CD33 Alzheimer’s Risk-Altering Polymorphism, CD33 Expression, and Exon 2 Splicing. J. Neurosci. 2013, 33, 13320–13325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s Disease Risk Gene CD33 Inhibits Microglial Uptake of Amyloid Beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, L.A.; Hermans, S.J.; Crespi, G.A.; Gooi, J.H.; Doughty, L.; Nero, T.L.; Markulić, J.; Ebneth, A.; Wroblowski, B.; Oehlrich, D.; et al. Small Molecule Binding to Alzheimer Risk Factor CD33 Promotes Aβ Phagocytosis. iScience 2019, 19, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Vom Berg, J.; Prokop, S.; Miller, K.R.; Obst, J.; Kälin, R.E.; Lopategui-Cabezas, I.; Wegner, A.; Mair, F.; Schipke, C.G.; Peters, O.; et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat. Med. 2012, 18, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Town, T.; Vendrame, M.; Patel, A.; Poetter, D.; DelleDonne, A.; Mori, T.; Smeed, R.; Crawford, F.; Klein, T.; Tan, J.; et al. Reduced Th1 and enhanced Th2 immunity after immunization with Alzheimer’s beta-amyloid(1-42). J. Neuroimmunol. 2002, 132, 49–59. [Google Scholar] [CrossRef]
- Hu, W.T.; Holtzman, D.M.; Fagan, A.M.; Shaw, L.M.; Perrin, R.; Arnold, S.E.; Grossman, M.; Xiong, C.; Craig-Schapiro, R.; Clark, C.M.; et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology 2012, 79, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.L.; Bowman, E.P.; McElwee, J.J.; Smyth, M.J.; Casanova, J.-L.; Cooper, A.M.; Cua, D.J. IL-12 and IL-23 cytokines: From discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 2015, 21, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Glatiramer acetate: A review of its use in patients with relapsing-remitting multiple sclerosis and in delaying the onset of clinically definite multiple sclerosis. CNS Drugs 2013, 27, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Arnon, R.; Aharoni, R. Glatiramer Acetate: From Bench to Bed and Back. ISR Med. Assoc. J. 2019, 21, 151–157. [Google Scholar] [PubMed]
- Chen, L.; Yao, Y.; Wei, C.; Sun, Y.; Ma, X.; Zhang, R.; Xu, X.; Hao, J. T cell immunity to glatiramer acetate ameliorates cognitive deficits induced by chronic cerebral hypoperfusion by modulating the microenvironment. Sci. Rep. 2015, 5, 14308. [Google Scholar] [CrossRef] [Green Version]
- Vieira, P.L.; Heystek, H.C.; Wormmeester, J.; Wierenga, E.A.; Kapsenberg, M.L. Glatiramer Acetate (Copolymer-1, Copaxone) Promotes Th2 Cell Development and Increased IL-10 Production Through Modulation of Dendritic Cells. J. Immunol. 2003, 170, 4483–4488. [Google Scholar] [CrossRef] [Green Version]
- Lalive, P.H.; Neuhaus, O.; Benkhoucha, M.; Burger, D.; Hohlfeld, R.; Zamvil, S.S.; Weber, M.S. Glatiramer acetate in the treatment of multiple sclerosis: Emerging concepts regarding its mechanism of action. CNS Drugs 2011, 25, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koronyo, Y.; Salumbides, B.C.; Sheyn, J.; Pelissier, L.; Li, S.; Ljubimov, V.; Moyseyev, M.; Daley, D.; Fuchs, D.-T.; Pham, M.; et al. Therapeutic effects of glatiramer acetate and grafted CD115+monocytes in a mouse model of Alzheimer’s disease. Brain 2015, 138, 2399–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butovsky, O.; Koronyo-Hamaoui, M.; Kunis, G.; Ophir, E.; Landa, G.; Cohen, H.; Schwartz, M. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc. Natl. Acad. Sci. USA 2006, 103, 11784–11789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frenkel, D.; Maron, R.; Burt, D.S.; Weiner, H.L. Nasal vaccination with a proteosome-based adjuvant and glatiramer acetate clears beta-amyloid in a mouse model of Alzheimer disease. J. Clin. Investig. 2005, 115, 2423–2433. [Google Scholar] [CrossRef] [Green Version]
- Bakalash, S.; Pham, M.; Koronyo, Y.; Salumbides, B.C.; Kramerov, A.; Seidenberg, H.; Berel, D.; Black, K.L.; Koronyo-Hamaoui, M. Egr1 Expression Is Induced Following Glatiramer Acetate Immunotherapy in Rodent Models of Glaucoma and Alzheimer’s Disease. Investig. Opthalmology Vis. Sci. 2011, 52, 9033–9046. [Google Scholar] [CrossRef]
- Khanna, A.K. MECHANISM OF THE COMBINATION IMMUNOSUPPRESSIVE EFFECTS OF RAPAMYCIN WITH EITHER CYCLOSPORINE OR TACROLIMUS. Transplantation 2000, 70, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.K.K.; Knapp, S.; Wurtman, R.J. Prostaglandin E2 Stimulates Amyloid Precursor Protein Gene Expression: Inhibition by Immunosuppressants. J. Neurosci. 1999, 19, 940–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojanathammanee, L.; Floden, A.M.; Manocha, G.D.; Combs, C.K. Attenuation of microglial activation in a mouse model of Alzheimer’s disease via NFAT inhibition. J. Neuroinflamm. 2015, 12, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozkalne, A.; Hyman, B.T.; Spires-Jones, T.L. Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol. Dis. 2011, 41, 650–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Singh, N. Calcineurin inhibitors improve memory loss and neuropathological changes in mouse model of dementia. Pharmacol. Biochem. Behav. 2017, 153, 147–159. [Google Scholar] [CrossRef]
- Alam, J.; Blackburn, K.; Patrick, D. Neflamapimod: Clinical Phase 2b-Ready Oral Small Molecule Inhibitor of p38α to Reverse Synaptic Dysfunction in Early Alzheimer’s Disease. J. Prev. Alzheimer's Dis. 2017, 4, 273–278. [Google Scholar] [PubMed]
- Alam, J.J. Selective Brain-Targeted Antagonism of p38 MAPKα Reduces Hippocampal IL-1β Levels and Improves Morris Water Maze Performance in Aged Rats. J. Alzheimer’s Dis. 2015, 48, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, M.I.; Ager, R.R.; Chu, S.-H.; Yazan, O.; Sanderson, S.D.; LaFerla, F.M.; Taylor, S.M.; Woodruff, T.M.; Tenner, A.J. Treatment with a C5aR Antagonist Decreases Pathology and Enhances Behavioral Performance in Murine Models of Alzheimer’s Disease. J. Immunol. 2009, 183, 1375–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lansita, J.A.; Mease, K.M.; Qiu, H.; Yednock, T.; Sankaranarayanan, S.; Kramer, S. Nonclinical Development of ANX005: A Humanized Anti-C1q Antibody for Treatment of Autoimmune and Neurodegenerative Diseases. Int. J. Toxicol. 2017, 36, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, M.-C.; Sawyer, N.; Greig, G.M.; Hamel, M.; Kargman, S.; Ducharme, Y.; Lau, C.K.; Friesen, R.W.; O’Neill, G.P.; Gervais, F.G.; et al. The C3a receptor antagonist SB 290157 has agonist activity. Immunol. Lett. 2005, 100, 139–145. [Google Scholar] [CrossRef]
- Porrini, V.; Lanzillotta, A.; Branca, C.; Benarese, M.; Parrella, E.; Lorenzini, L.; Calza, L.; Flaibani, R.; Spano, P.; Imbimbo, B.; et al. CHF5074 (CSP-1103) induces microglia alternative activation in plaque-free Tg2576 mice and primary glial cultures exposed to beta-amyloid. Neuroscience 2015, 302, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Hwangbo, D.-S.; Lee, H.-Y.; Abozaid, L.S.; Min, K.-J. Mechanisms of Lifespan Regulation by Calorie Restriction and Intermittent Fasting in Model Organisms. Nutrients 2020, 12, 1194. [Google Scholar] [CrossRef] [PubMed]
- Dorling, J.L.; Martin, C.K.; Redman, L.M. Calorie restriction for enhanced longevity: The role of novel dietary strategies in the present obesogenic environment. Ageing Res. Rev. 2020, 64, 101038. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Kim, H.J.; Kim, J.W.; Yu, B.P. The inflammation hypothesis of aging: Molecular modulation by calorie restriction. Ann. N. Y. Acad. Sci. 2001, 928, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, A.; Di Germanio, C.; Bernier, M.; de Cabo, R. A time to fast. Science 2018, 362, 770–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-W.; He, S.-J.; Feng, X.; Cheng, J.; Luo, Y.-T.; Tian, L.; Huang, Q. Metformin: A review of its potential indications. Drug Des. Dev. Ther. 2017, ume 11, 2421–2429. [Google Scholar] [CrossRef] [Green Version]
- Vieira, R.; Souto, S.B.; Sánchez-López, E.; Machado, A.L.; Severino, P.; Jose, S.; Santini, A.; Fortuna, A.; García, M.L.; Silva, A.M. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome—Review of Classical and New Compounds: Part-I. Pharmaceuticals 2019, 12, 152. [Google Scholar] [CrossRef] [Green Version]
- Partridge, L.; Piper, M.D.; Mair, W. Dietary restriction in Drosophila. Mech. Ageing Dev. 2005, 126, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Masoro, E.J. Caloric restriction and aging: An update. Exp. Gerontol. 2000, 35, 299–305. [Google Scholar] [CrossRef]
- Onken, B.; Driscoll, M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 2010, 5, e8758. [Google Scholar] [CrossRef]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55.e6. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.B.; Koo, J.H.; Jang, Y.C.; Yang, C.H.; Lee, Y.; Cosio-Lima, L.M.; Cho, J.Y. Neuroprotective Effects of Endurance Exercise Against High-Fat Diet-Induced Hippocampal Neuroinflammation. J. Neuroendocrinol. 2016, 28. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Cheon, W.; Kim, K. Effects of Long-Term Endurance Exercise and Lithium Treatment on Neuroprotective Factors in Hippocampus of Obese Rats. Int. J. Environ. Res. Public Heal. 2020, 17, 3317. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Lu, J.; Rodova, M.; Lezi, E.; Crafter, A.B.; Swerdlow, R.H. Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 228–234. [Google Scholar] [CrossRef] [Green Version]
- Radak, Z.; Suzuki, K.; Higuchi, M.; Balogh, L.; Boldogh, I.; Koltai, E. Physical exercise, reactive oxygen species and neuroprotection. Free Radic. Biol. Med. 2016, 98, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, K.A.; Cotman, C.W. Exercise counteracts declining hippocampal function in aging and Alzheimer’s disease. Neurobiol. Dis. 2013, 57, 47–55. [Google Scholar] [CrossRef]
- Dalle, S.; Rossmeislova, L.; Koppo, K. The Role of Inflammation in Age-Related Sarcopenia. Front. Physiol. 2017, 8, 1045. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, M.I.; Bourgeois, J.M.; Nederveen, J.P.; Leite, M.R.; Hettinga, B.P.; Bujak, A.L.; May, L.; Lin, E.; Crozier, M.; Rusiecki, D.R.; et al. Lifelong aerobic exercise protects against inflammaging and cancer. PLoS ONE 2019, 14, e0210863. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and inflammation-Story of a double-edged blade. J. Pineal Res. 2018, 65, e12525. [Google Scholar] [CrossRef] [Green Version]
- Hardeland, R. Aging, Melatonin, and the Pro- and Anti-Inflammatory Networks. Int. J. Mol. Sci. 2019, 20, 1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchal, J.; Pifferi, F.; Aujard, F. Resveratrol in mammals: Effects on aging biomarkers, age-related diseases, and life span. Ann. N. Y. Acad. Sci. 2013, 1290, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Alcaín, F.J.; Villalba, J.M. Sirtuin activators. Expert. Opin. Ther. Pat. 2009, 19, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Liu, Q.; Wang, M.; Liang, M.; Yang, X.; Xu, X.; Zou, H.; Qiu, J. Activation of Sirt1 by Resveratrol Inhibits TNF-α Induced Inflammation in Fibroblasts. PLoS ONE 2011, 6, e27081. [Google Scholar] [CrossRef]
- Arbo, B.D.; André-Miral, C.; Nasre-Nasser, R.G.; Schimith, L.E.; Santos, M.G.; Costa-Silva, D.; Muccillo-Baisch, A.L.; Hort, M.A. Resveratrol Derivatives as Potential Treatments for Alzheimer’s and Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 103. [Google Scholar] [CrossRef]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, R.S.; Thomas, R.G.; Craft, S.; Van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S.; et al. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef]
- Witte, A.V.; Kerti, L.; Margulies, D.S.; Flöel, A. Effects of resveratrol on memory performance, hippocampal functional connectivity, and glucose metabolism in healthy older adults. J. Neurosci. 2014, 34, 7862–7870. [Google Scholar] [CrossRef] [Green Version]
- Jiao, F.; Gong, Z. The Beneficial Roles of SIRT1 in Neuroinflammation-Related Diseases. Oxid. Med. Cell Longev. 2020, 2020, 6782872. [Google Scholar] [CrossRef] [PubMed]
- Cannizzo, E.S.; Clement, C.C.; Sahu, R.; Follo, C.; Santambrogio, L. Oxidative stress, inflamm-aging and immunosenescence. J. Proteom. 2011, 74, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.C.; Bellalta, S.; Basoalto, R.; Gómez-Valenzuela, F.; Jalil, Y.; Lépez, M.; Matamoros, A.; Von Bernhardi, R. The Challenge by Multiple Environmental and Biological Factors Induce Inflammation in Aging: Their Role in the Promotion of Chronic Disease. Front. Immunol. 2020, 11, 570083. [Google Scholar] [CrossRef]
- Chelombitko, M.A. Role of Reactive Oxygen Species in Inflammation: A Minireview. Mosc. Univ. Biol. Sci. Bull. 2018, 73, 199–202. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Yin, J.; Chen, J.; Ma, X.; Wu, M.; Liu, G.; Yao, K.; Tan, B.; Yin, Y. Mitochondria-Targeted Antioxidants: A Step towards Disease Treatment. Oxidative Med. Cell. Longev. 2020, 2020, 1–18. [Google Scholar] [CrossRef]
- Fujimoto, C.; Yamasoba, T. Mitochondria-Targeted Antioxidants for Treatment of Hearing Loss: A Systematic Review. Antioxidants 2019, 8, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Hu, X.H.; Jia, Z.G.; Xu, M.H.; Guo, Z.Y.; Gao, F.H. Tiron protects against UVB-induced senescence-like characteristics in human dermal fibroblasts by the inhibition of superoxide anion production and glutathione depletion. Australas. J. Dermatol. 2012, 53, 172–180. [Google Scholar] [CrossRef]
- For The Carmis Study Group; Piermarocchi, S.; Saviano, S.; Parisi, V.; Tedeschi, M.; Panozzo, G.; Scarpa, G.; Boschi, G.; Giudice, G.L.; Sartore, M.; et al. Carotenoids in Age-Related Maculopathy Italian Study (CARMIS): Two-Year Results of a Randomized Study. Eur. J. Ophthalmol. 2012, 22, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, X.; Xiang, Q.; Meng, X.; Peng, Y.; Du, N.; Liu, Z.; Sun, Q.; Wang, C.; Liu, X. Astaxanthin alleviates brain aging in rats by attenuating oxidative stress and increasing BDNF levels. Food Funct. 2013, 5, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.S.; Rutherfurd, K.J.; Cross, M.L.; Gopal, P.K. Enhancement of immunity in the elderly by dietary supplementation with the probiotic Bifidobacterium lactis HN019. Am. J. Clin. Nutr. 2001, 74, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Rowland, I.; Thomas, L.V.; Yaqoob, P. Immunomodulatory effects of a probiotic drink containing Lactobacillus casei Shirota in healthy older volunteers. Eur. J. Nutr. 2013, 52, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Vulevic, J.; Drakoularakou, A.; Yaqoob, P.; Tzortzis, G.; Gibson, G.R. Modulation of the fecal microflora profile and immune function by a novel trans-galactooligosaccharide mixture (B-GOS) in healthy elderly volunteers. Am. J. Clin. Nutr. 2008, 88, 1438–1446. [Google Scholar] [PubMed]
- Frasca, D.; Blomberg, B.B. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 2016, 17, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Man, A.W.C.; Zhou, Y.; Xia, N.; Li, H. Involvement of Gut Microbiota, Microbial Metabolites and Interaction with Polyphenol in Host Immunometabolism. Nutrients 2020, 12, 3054. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Cechova, K.; Amlerova, J.; Hort, J. Antibiotics, gut microbiota, and Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 1–10. [Google Scholar] [CrossRef]
- He, Y.; Li, B.; Sun, D.; Chen, S. Gut Microbiota: Implications in Alzheimer’s Disease. J. Clin. Med. 2020, 9, 2042. [Google Scholar] [CrossRef] [PubMed]
- Haran, J.P.; Bhattarai, S.K.; Foley, S.E.; Dutta, P.; Ward, D.V.; Bucci, V.; McCormick, B.A. Alzheimer’s Disease Microbiome Is Associated with Dysregulation of the Anti-Inflammatory P-Glycoprotein Pathway. mBio 2019, 10, e00632-19. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.; Li, C.; Chu, F.; Tian, X.; Zhu, J. Target Dysbiosis of Gut Microbes as a Future Therapeutic Manipulation in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 544235. [Google Scholar] [CrossRef]
- Sharma, R.; Padwad, Y. Probiotic bacteria as modulators of cellular senescence: Emerging concepts and opportunities. Gut Microbes 2020, 11, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, U.; Arshad, M.; Sameen, A.; Oh, D.-H. Crosstalk between Gut and Brain in Alzheimer’s Disease: The Role of Gut Microbiota Modulation Strategies. Nutrients 2021, 13, 690. [Google Scholar] [CrossRef] [PubMed]
- Kesika, P.; Suganthy, N.; Sivamaruthi, B.S.; Chaiyasut, C. Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer’s disease. Life Sci. 2021, 264, 118627. [Google Scholar] [CrossRef] [PubMed]
- Bonfili, L.; Cecarini, V.; Gogoi, O.; Gong, C.; Cuccioloni, M.; Angeletti, M.; Rossi, G.; Eleuteri, A.M. Microbiota modulation as preventative and therapeutic approach in Alzheimer’s disease. FEBS J. 2020. [Google Scholar] [CrossRef]
- Liu, S.; Gao, J.; Zhu, M.; Liu, K.; Zhang, H.-L. Gut Microbiota and Dysbiosis in Alzheimer’s Disease: Implications for Pathogenesis and Treatment. Mol. Neurobiol. 2020, 57, 5026–5043. [Google Scholar] [CrossRef] [PubMed]
- Portis, S.M.; Chaput, D.; Burroughs, B.; Hudson, C.; Sanberg, P.R.; Bickford, P.C. Effects of nutraceutical intervention on serum proteins in aged rats. GeroScience 2020, 42, 703–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadhukhan, P.; Saha, S.; Dutta, S.; Mahalanobish, S.; Sil, P.C. Nutraceuticals: An emerging therapeutic approach against the pathogenesis of Alzheimer’s disease. Pharmacol. Res. 2018, 129, 100–114. [Google Scholar] [CrossRef]
- Calfio, C.; Gonzalez, A.; Singh, S.K.; Rojo, L.E.; Maccioni, R.B. The Emerging Role of Nutraceuticals and Phytochemicals in the Prevention and Treatment of Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 77, 33–51. [Google Scholar] [CrossRef]
- Vaiserman, A.; Koliada, A.; Lushchak, O. Neuroinflammation in pathogenesis of Alzheimer’s disease: Phytochemicals as potential therapeutics. Mech. Ageing Dev. 2020, 189, 111259. [Google Scholar] [CrossRef] [PubMed]
- Flowers, A.; Lee, J.-Y.; Acosta, S.; Hudson, C.; Small, B.; Sanberg, C.D.; Bickford, P.C.; Grimmig, B. NT-020 treatment reduces inflammation and augments Nrf-2 and Wnt signaling in aged rats. J. Neuroinflamm. 2015, 12, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpert, P.T. The Role of Vitamins and Minerals on the Immune System. Home Heal. Care Manag. Pr. 2017, 29, 199–202. [Google Scholar] [CrossRef]
- Mora, J.R.; Iwata, M.; von Andrian, U.H. Vitamin effects on the immune system: Vitamins A and D take centre stage. Nat. Rev. Immunol. 2008, 8, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggini, S.; Pierre, A.; Calder, P.C. Immune Function and Micronutrient Requirements Change over the Life Course. Nutrients 2018, 10, 491. [Google Scholar]
- Abiri, B.; Vafa, M. Micronutrients that Affect Immunosenescence. Adv. Exp. Med. Biol. 2020, 1260, 13–31. [Google Scholar]
- Meydani, S.N.; Meydani, M.; Blumberg, J.B.; Leka, L.S.; Siber, G.; Loszewski, R.; Thompson, C.; Pedrosa, M.C.; Diamond, R.D.; Stollar, B.D. Vitamin E supplementation and in vivo immune response in healthy elderly subjects. A randomized controlled trial. JAMA 1997, 277, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.; Hernanz, A.; Guayerbas, N.; Victor, V.M.; Arnalich, F. Vitamin E ingestion improves several immune functions in elderly men and women. Free Radic Res. 2008, 42, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Pallast, E.G.; Schouten, E.G.; De Waart, F.G.; Fonk, H.C.; Doekes, G.; Von Blomberg, B.M.; Kok, F.J. Effect of 50- and 100-mg vitamin E supplements on cellular immune function in noninstitutionalized elderly persons. Am. J. Clin. Nutr. 1999, 69, 1273–1281. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.-P.-P.; Rahman, H.S. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, B.L.; Norhaizan, M.E. Carotenoids: How Effective Are They to Prevent Age-Related Diseases? Molecules 2019, 24, 1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monacelli, F.; Acquarone, E.; Giannotti, C.; Borghi, R.; Nencioni, A. Vitamin C, Aging and Alzheimer’s Disease. Nutrients 2017, 9, 670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggini, S.; Wintergerst, E.S.; Beveridge, S.; Hornig, D.H. Selected vitamins and trace elements support immune function by strengthening epithelial barriers and cellular and humoral immune responses. Br. J. Nutr. 2007, 98, S29–S35. [Google Scholar] [CrossRef] [PubMed]
- Zhitkovich, A. Nuclear and Cytoplasmic Functions of Vitamin C. Chem. Res. Toxicol. 2020, 33, 2515–2526. [Google Scholar] [CrossRef]
- Hemilae, H. Vitamin C and infections. Nutrients 2017, 9, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, A.C.; Maggini, S. Vitamin C and Immune Function. Nutrients 2017, 9, 1211. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Muto, N.; Gohda, E.; Yamamoto, I. Enhancement by Ascorbic Acid 2-Glucoside or Repeated Additions of Ascorbate of Mitogen-Induced IgM and IgG Productions by Human Peripheral Blood Lymphocytes. Jpn. J. Pharmacol. 1994, 66, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, Á.J.R. Zinc, aging, and immunosenescence: An overview. Pathobiol. Aging Age-Related Dis. 2015, 5, 25592. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, M.; Olbert, M.; Wyszogrodzka, G.; Młyniec, K.; Librowski, T. Antioxidant and anti-inflammatory effects of zinc. Zinc-dependent NF-κB signaling. Inflammopharmacology 2017, 25, 11–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.P.; Ho, E. Zinc and its role in age-related inflammation and immune dysfunction. Mol. Nutr. Food Res. 2012, 56, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S. Clinical, immunological, anti-inflammatory and antioxidant roles of zinc. Exp. Gerontol. 2008, 43, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Fortes, C.; Forastiere, F.; Agabiti, N.; Fano, V.; Pacifici, R.; Virgili, F.; Piras, G.; Guidi, L.; Bartoloni, C.; Tricerri, A.; et al. The Effect of Zinc and Vitamin A Supplementation on Immune Response in an Older Population. J. Am. Geriatr. Soc. 1998, 46, 19–26. [Google Scholar] [CrossRef]
- Lee, S.R. Critical role of zinc as either an antioxidant or a prooxidant in cellular systems. Oxid. Med. Cell Longev. 2018, 2018, 9156285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecora, F.; Persico, F.; Argentiero, A.; Neglia, C.; Esposito, S. The Role of Micronutrients in Support of the Immune Response against Viral Infections. Nutrients 2020, 12, 3198. [Google Scholar] [CrossRef]
- Prasad, A.S.; Bao, B.; Beck, F.W.; Kucuk, O.; Sarkar, F.H. Antioxidant effect of zinc in humans. Free. Radic. Biol. Med. 2004, 37, 1182–1190. [Google Scholar] [CrossRef]
- Xia, S.; Shen, Y.; Yu, W. Study on mechanism and intervention of inflamm-aging in rats. Chin. J. Gerontol. 2009, 29, 2595–2598. [Google Scholar]
- Xia, S.; Shen, Y.; Yu, W. Inflamm-aging related genes expression in aged rat hippocampus and Icariin intervention outcome. Geriatr. Health Care 2008, 14, 340–344. [Google Scholar]
- Zheng, J.; Hu, S.; Wang, J.; Zhang, X.; Yuan, D.; Zhang, C.; Liu, C.; Wang, T.; Zhou, Z. Icariin improves brain function decline in aging rats by enhancing neuronal autophagy through the AMPK/mTOR/ULK1 pathway. Pharm. Biol. 2021, 59, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-X.; Deng, Y.-Y.; Li, F.; Liu, B.; Liu, H.-Y.; Shi, J.-S.; Gong, Q.-H. Icariin, a major constituent of flavonoids from Epimedium brevicornum, protects against cognitive deficits induced by chronic brain hypoperfusion via its anti-amyloidogenic effect in rats. Pharmacol. Biochem. Behav. 2015, 138, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Wen, D.S.; Zhao, Y.R.; Xu, S.J. Epimedium sagittatum inhibits TLR4/MD-2 mediated NF-κB signaling pathway with anti-inflammatory activity. BMC Complement. Altern. Med. 2018, 18, 303. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Zhang, C.; Qian, C.; Na, M.; Ding, Y.; Zhao, H. Total Flavonoids of Epimedium Reduce Inflammatory Reaction via AMPK/SIRT1/NFκB Signaling Pathway in Testes of Natural Aging Rats. Nat. Prod. Res. Dev. 2018, 30, 1489–1493. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onyango, I.G.; Jauregui, G.V.; Čarná, M.; Bennett, J.P., Jr.; Stokin, G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. https://doi.org/10.3390/biomedicines9050524
Onyango IG, Jauregui GV, Čarná M, Bennett JP Jr., Stokin GB. Neuroinflammation in Alzheimer’s Disease. Biomedicines. 2021; 9(5):524. https://doi.org/10.3390/biomedicines9050524
Chicago/Turabian StyleOnyango, Isaac G., Gretsen V. Jauregui, Mária Čarná, James P. Bennett, Jr., and Gorazd B. Stokin. 2021. "Neuroinflammation in Alzheimer’s Disease" Biomedicines 9, no. 5: 524. https://doi.org/10.3390/biomedicines9050524