
Characterization and Full Genome Sequence of Novel KPP-5 Lytic Phage against Klebsiella pneumoniae Responsible for Recalcitrant Infection

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strain for Bacteriophage Isolation
2.2. Inoculum Preparation of the Host Isolate
2.3. Bacteriophage Isolation and Purification
2.4. Host Range Determination
2.5. Transmission Electron Microscopy (TEM)
2.6. Determination of KPP-5 Phage Bacterial Culture Clearance Tendency
2.7. One-Step Growth Curve
2.8. Assessment of KPP-5 phage Thermal and pH Stability
2.9. Phage DNA Isolation and Genome Sequencing of KPP-5 Phage
2.10. Bioinformatic Analysis of KPP-5 Phage Genome
3. Results and Discussion
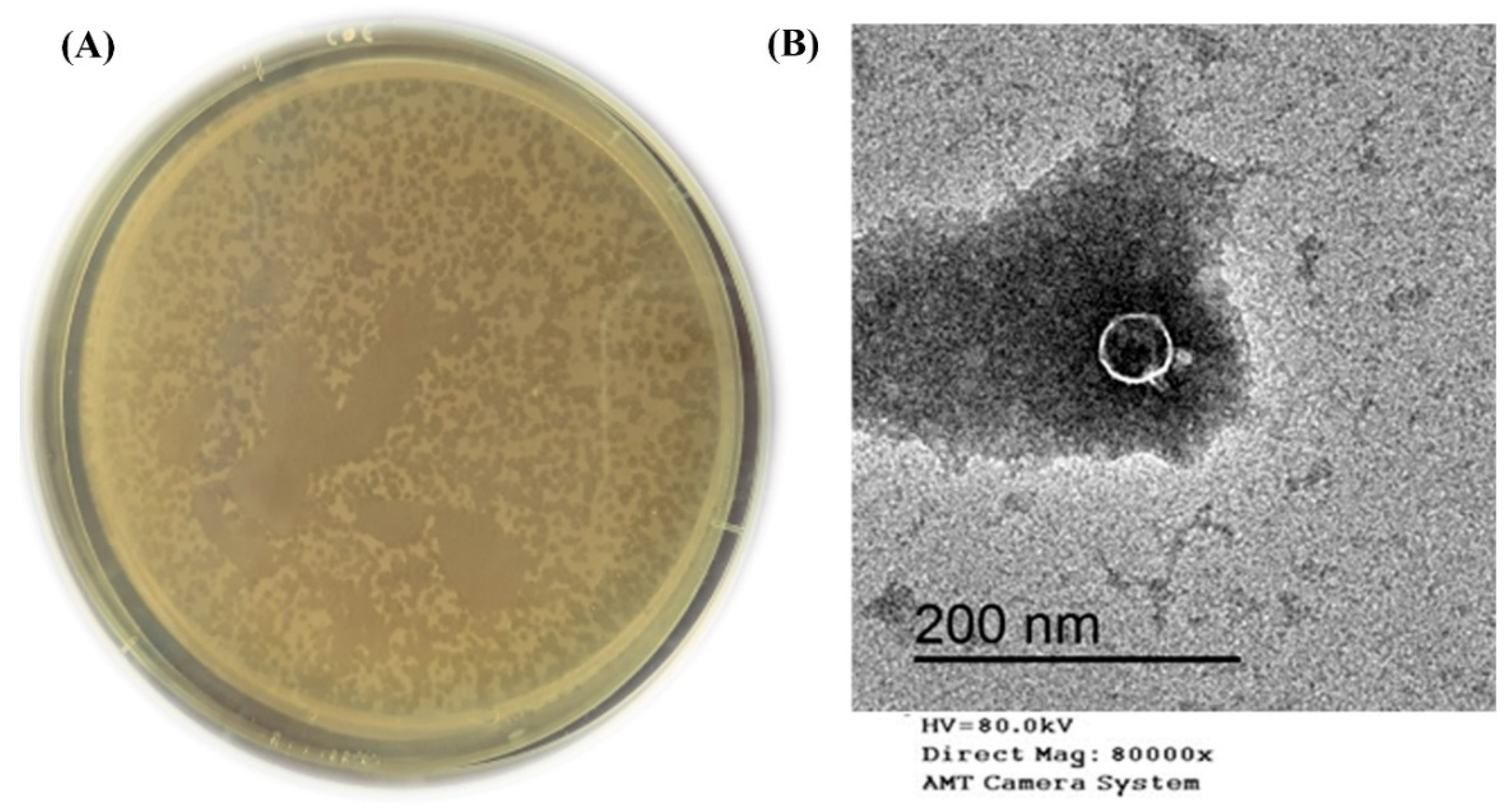
3.1. Broad Host Range Bacteriophage Isolation and Morphology
3.2. One-Step Growth Curve and Thermal and pH Stability of KPP-5 Phage
3.3. Genomic Analysis and Annotation of the KPP-5 Phage
3.4. Comparative Genomic Analysis
3.5. Specific Features of the KPP-5 Phage Genome
3.5.1. Replication, Regulation, Transcription, and Translation-Related Genes
3.5.2. Host Cell Lysis Related Genes
3.5.3. Phage Structure Related Genes
3.5.4. DNA Packaging-Related Genes
3.5.5. Related Unknown Function Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harada, S.; Aoki, K.; Yamamoto, S.; Ishii, Y.; Sekiya, N.; Kurai, H.; Furukawa, K.; Doi, A.; Tochitani, K.; Kubo, K.; et al. Clinical and molecular characteristics of Klebsiella pneumoniae isolates causing bloodstream infections in Japan: Occurrence of hypervirulent infections in health care. J. Clin. Microbiol. 2019, 57, e01206-19. [Google Scholar] [CrossRef] [Green Version]
- Neyrolles, O.; Brisse, S.; Fevre, C.; Passet, V.; Issenhuth-Jeanjean, S.; Tournebize, R.; Diancourt, L.; Grimont, P. Virulent clones of Klebsiella pneumoniae: Identification and evolutionary scenario based on genomic and phenotypic characterization. PLoS ONE 2009, 4, e4982. [Google Scholar] [CrossRef] [Green Version]
- Cabral, A.B.; Melo, R.d.C.d.A.; Maciel, M.A.V.; Lopes, A.C.S. Multidrug resistance genes, including blaKPC and blaCTX-M-2, among Klebsiella pneumoniae isolated in Recife, Brazil. Rev. Da Soc. Bras. De Med. Trop. 2012, 45, 572–578. [Google Scholar] [CrossRef]
- Overdevest, I.T.M.A.; Heck, M.; van der Zwaluw, K.; Huijsdens, X.; van Santen, M.; Rijnsburger, M.; Eustace, A.; Xu, L.; Hawkey, P.; Savelkoul, P.; et al. Extended-spectrum β-lactamase producing Klebsiella spp. in chicken meat and humans: A comparison of typing methods. Clin. Microbiol. Infect. 2014, 20, 251–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-S.; Chon, J.-W.; Kim, Y.-J.; Kim, D.-H.; Kim, M.-s.; Seo, K.-H. Prevalence and characterization of extended-spectrum-β-lactamase-producing Escherichia coli and Klebsiella pneumoniae in ready-to-eat vegetables. Int. J. Food Microbiol. 2015, 207, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.S.; Price, L.B. Recent research examining links among Klebsiella pneumoniae from food, food animals, and human extraintestinal infections. Curr. Environ. Health Rep. 2016, 3, 128–135. [Google Scholar] [CrossRef]
- Holt, K.E.; Wertheim, H.; Zadoks, R.N.; Baker, S.; Whitehouse, C.A.; Dance, D.; Jenney, A.; Connor, T.R.; Hsu, L.Y.; Severin, J.; et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl. Acad. Sci. USA 2015, 112, E3574–E3581. [Google Scholar] [CrossRef] [Green Version]
- Flemming, H.-C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An emergent form of bacterial life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Sofy, A.R.; Aboseidah, A.A.; El-Morsi, E.-S.; Azmy, H.A.; Hmed, A.A. Evaluation of antibacterial and antibiofilm activity of new antimicrobials as an urgent need to counteract stubborn multidrug-resistant bacteria. J. Pure Appl. Microbiol. 2020, 14, 595–608. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, S.M.; Cardoso, M.H.; Cândido, E.d.S.; Franco, O.L. Understanding, preventing and eradicating Klebsiella pneumoniae biofilms. Future Microbiol. 2016, 11, 527–538. [Google Scholar] [CrossRef]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage therapy: A renewed spproach to combat antibiotic-resistant bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef] [Green Version]
- Gordillo Altamirano, F.L.; Barr, J.J. Phage therapy in the postantibiotic era. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef] [Green Version]
- Abedon, S. Phage therapy pharmacology: Calculating phage dosing. Adv. Appl. Microbiol. 2011, 77, 1–40. [Google Scholar] [CrossRef]
- Elshaarawy, R.F.M.; Mustafa, F.H.A.; Sofy, A.R.; Hmed, A.A.; Janiak, C. A new synthetic antifouling coatings integrated novel aminothiazole-functionalized ionic liquids motifs with enhanced antibacterial performance. J. Environ. Chem. Eng. 2019, 7. [Google Scholar] [CrossRef]
- Domingo-Calap, P.; Georgel, P.; Bahram, S. Back to the future: Bacteriophages as promising therapeutic tools. Hla 2016, 87, 133–140. [Google Scholar] [CrossRef]
- Heilmann, S.; Sneppen, K.; Krishna, S. Coexistence of phage and bacteria on the boundary of self-organized refuges. Proc. Natl. Acad. Sci. USA 2012, 109, 12828–12833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.P.; Oliveira, H.; Melo, L.D.; Sillankorva, S.; Azeredo, J. Bacteriophage-encoded depolymerases: Their diversity and biotechnological applications. Appl. Microbiol. Biotechnol. 2016, 100, 2141–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spruit, C.M.; Wicklund, A.; Wan, X.; Skurnik, M.; Pajunen, M.I. Discovery of three toxic proteins of Klebsiella Phage fHe-Kpn01. Viruses 2020, 12, 544. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.R.; Donovan, D.M. Antimicrobial bacteriophage-derived proteins and therapeutic applications. Bacteriophage 2015, 5, e1062590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roucourt, B.; Lavigne, R. The role of interactions between phage and bacterial proteins within the infected cell: A diverse and puzzling interactome. Environ. Microbiol. 2009, 11, 2789–2805. [Google Scholar] [CrossRef]
- Funke, G.; Funke-Kissling, P. Evaluation of the new VITEK 2 card for identification of clinically relevant gram-negative rods. J. Clin. Microbiol. 2004, 42, 4067–4071. [Google Scholar] [CrossRef] [Green Version]
- Funke, G.; Funke-Kissling, P. Performance of the new VITEK 2 GP card for identification of medically relevant gram-positive Cocci in a routine clinical laboratory. J. Clin. Microbiol. 2005, 43, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Bauer, A.W.; Kirby, W.M.; Sherris, J.C.; Turck, M. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 1966, 45, 493–496. [Google Scholar] [CrossRef] [PubMed]
- NCCLS/CLSI. Performance Standards for Antimicrobial Susceptibility Testing; National Committee for Clinical Laboratory Standards/Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2007. [Google Scholar]
- Sieuwerts, S.; de Bok, F.A.; Mols, E.; de vos, W.M.; Vlieg, J.E. A simple and fast method for determining colony forming units. Lett. Appl. Microbiol. 2008, 47, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Bibi, Z.; Abbas, Z.; Rehman, S. The phage P.E1isolated from hospital sewage reduces the growth of Escherichia coli. Biocontrol. Sci. Technol. 2015, 26, 181–188. [Google Scholar] [CrossRef]
- Sangha, K.K.; Kumar, B.V.; Agrawal, R.K.; Deka, D.; Verma, R. Proteomic characterization of lytic bacteriophages of Staphylococcus aureus isolated from sewage affluent of India. Int. Sch. Res. Not. 2014, 2014, 265298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Shi, J.; Ma, W.; Li, Z.; Wang, J.; Li, J.; Wang, X. Isolation, characterization, and application of a novel specific Salmonella bacteriophage in different food matrices. Food Res. Int. 2018, 111, 631–641. [Google Scholar] [CrossRef]
- Jun, J.W.; Kim, J.H.; Shin, S.P.; Han, J.E.; Chai, J.Y.; Park, S.C. Protective effects of the Aeromonas phages pAh1-C and pAh6-C against mass mortality of the cyprinid loach (Misgurnus anguillicaudatus) caused by Aeromonas hydrophila. Aquaculture 2013, 416–417, 289–295. [Google Scholar] [CrossRef]
- Mirzaei, M.K.; Nilsson, A.S. Correction: Isolation of phages for phage therapy: A comparison of spot tests and efficiency of plating analyses for determination of host range and efficacy. PLoS ONE 2015, 10, e0127606. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, H.W. Bacteriophage electron microscopy. Adv. Virus Res. 2012, 82, 1–32. [Google Scholar] [CrossRef]
- Accolas, J.P.; Spillmann, H. The morphology of six bacteriophages of Streptococcus thermophilus. J. Appl. Bacteriol. 1979, 47, 135–144. [Google Scholar] [CrossRef]
- Khawaja, K.A.; Rauf, M.; Abbas, Z.; Rehman, S. A virulent phage JHP against Pseudomonas aeruginosa showed infectivity against multiple genera. J. Basic Microbiol. 2016, 56, 1090–1097. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Lv, Y.; Zheng, W.; Mi, Z.; Pei, G.; An, X.; Xu, X.; Han, C.; Liu, J.; et al. Characterization and complete genome sequence analysis of novel bacteriophage IME-EFm1 infecting Enterococcus faecium. J. Gen. Virol. 2014, 95, 2565–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philipson, L.; Albertsson, P.A.; Frick, G. The purification and concentration of viruses by aqueous polymerphase systems. Virology 1960, 11, 553–571. [Google Scholar] [CrossRef]
- Jamalludeen, N.; Johnson, R.P.; Friendship, R.; Kropinski, A.M.; Lingohr, E.J.; Gyles, C.L. Isolation and characterization of nine bacteriophages that lyse O149 enterotoxigenic Escherichia coli. Vet. Microbiol. 2007, 124, 47–57. [Google Scholar] [CrossRef]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2011, 40, D290–D301. [Google Scholar] [CrossRef]
- Conant, G.C.; Wolfe, K.H. GenomeVx: Simple web-based creation of editable circular chromosome maps. Bioinformatics 2008, 24, 861–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Kleinheinz, K.A.; Joensen, K.G.; Larsen, M.V. Applying the ResFinder and VirulenceFinder web-services for easy identification of acquired antibiotic resistance and E. coli virulence genes in bacteriophage and prophage nucleotide sequences. Bacteriophage 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darzentas, N. Circoletto: Visualizing sequence similarity with Circos. Bioinformatics 2010, 26, 2620–2621. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, P.; King, J.F.; Seto, D. CGUG: In silico proteome and genome parsing tool for the determination of “core” and unique genes in the analysis of genomes up to ca. 1.9 Mb. BMC Res. Notes 2009, 2. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Göker, M.; Kelso, J. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Xu, X.; Zhang, Z.; Shen, H.; Chen, J.; Zhang, K. Molecular characterization of clinical multidrug-resistant Klebsiella pneumoniae isolates. Ann. Clin. Microbiol. Antimicrob. 2014, 13, 16. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Wang, S.; Zhan, L.; Jin, Y.; Duan, J.; Hao, Z.; Lv, J.; Qi, X.; Chen, L.; Kreiswirth, B.N.; et al. Microbiological and clinical characteristics of hypermucoviscous Klebsiella pneumoniae isolates associated with invasive infections in China. Front. Cell. Infect. Microbiol. 2017, 7, 24. [Google Scholar] [CrossRef]
- Chung, P.Y.; Jass, J. The emerging problems of Klebsiella pneumoniae infections: Carbapenem resistance and biofilm formation. FEMS Microbiol. Lett. 2016, 363, fnw219. [Google Scholar] [CrossRef] [Green Version]
- Woźniakowski, G.; Kuralayanapalya, S.P.; Patil, S.S.; Hamsapriya, S.; Shinduja, R.; Roy, P.; Amachawadi, R.G. Prevalence of extended-spectrum beta-lactamase producing bacteria from animal origin: A systematic review and meta-analysis report from India. PLoS ONE 2019, 14, e0221771. [Google Scholar] [CrossRef]
- Lai, Y.-C.; Lu, M.-C.; Hsueh, P.-R. Hypervirulence and carbapenem resistance: Two distinct evolutionary directions that led high-risk Klebsiella pneumoniae clones to epidemic success. Expert Rev. Mol. Diagn. 2019, 19, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Sofy, A.R.; Abd El Haliem, N.F.; Refaey, E.E.; Hmed, A.A. Polyvalent phage CoNShP-3 as a natural antimicrobial agent showing lytic and antibiofilm activities against antibiotic-resistant coagulase-negative staphylococci strains. Foods 2020, 9, 673. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, M.M.; Marmo, P.; Henrici De Angelis, L.; Palmieri, M.; Ciacci, N.; Di Lallo, G.; Demattè, E.; Vannuccini, E.; Lupetti, P.; Rossolini, G.M.; et al. φBO1E, a newly discovered lytic bacteriophage targeting carbapenemase-producing Klebsiella pneumoniae of the pandemic Clonal Group 258 clade II lineage. Sci. Rep. 2017, 7, 2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drulis-Kawa, Z.; Majkowska-Skrobek, G.; Maciejewska, B. Bacteriophages and phage-derived proteins—Application approaches. Curr. Med. Chem. 2015, 22, 1757–1773. [Google Scholar] [CrossRef]
- Cao, F.; Wang, X.; Wang, L.; Li, Z.; Che, J.; Wang, L.; Li, X.; Cao, Z.; Zhang, J.; Jin, L.; et al. Evaluation of the efficacy of a bacteriophage in the treatment of pneumonia induced by multidrug resistance Klebsiella pneumoniaein mice. BioMed Res. Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hyman, P.; Abedon, S.T. Bacteriophage host range and bacterial resistance. Adv. Appl. Microbiol. 2010, 70, 217–248. [Google Scholar]
- Lin, T.-L.; Hsieh, P.-F.; Huang, Y.-T.; Lee, W.-C.; Tsai, Y.-T.; Su, P.-A.; Pan, Y.-J.; Hsu, C.-R.; Wu, M.-C.; Wang, J.-T. Isolation of a bacteriophage and its depolymerase specific for K1 capsule of Klebsiella pneumoniae: Implication in typing and treatment. J. Infect. Dis. 2014, 210, 1734–1744. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.-J.; Lin, T.-L.; Chen, C.-C.; Tsai, Y.-T.; Cheng, Y.-H.; Chen, Y.-Y.; Hsieh, P.-F.; Lin, Y.-T.; Wang, J.-T.; Sandri-Goldin, R.M. Klebsiella phage ΦK64-1 encodes multiple depolymerases for multiple host capsular types. J. Virol. 2017, 91, e02457-16. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.; Zhang, Y.; Cheng, M.; Le, S.; Gu, J.; Bao, J.; Qin, J.; Guo, X.; Zhu, T. Characterization of Klebsiella pneumoniae ST11 isolates and their interactions with lytic phages. Viruses 2019, 11, 1080. [Google Scholar] [CrossRef] [Green Version]
- Lavigne, R.; Darius, P.; Summer, E.J.; Seto, D.; Mahadevan, P.; Nilsson, A.S.; Ackermann, H.W.; Kropinski, A.M. Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiol. 2009, 9, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitbart, M.; Miyake, J.H.; Rohwer, F. Global distribution of nearly identical phage-encoded DNA sequences. FEMS Microbiol. Lett. 2004, 236, 249–256. [Google Scholar] [CrossRef]
- Jamal, M.; Hussain, T.; Das, C.R.; Andleeb, S. Characterization of Siphoviridae phage Z and studying its efficacy against multidrug-resistant Klebsiella pneumoniae planktonic cells and biofilm. J. Med. Microbiol. 2015, 64, 454–462. [Google Scholar] [CrossRef] [Green Version]
- Tabassum, R.; Shafique, M.; Khawaja, K.A.; Alvi, I.A.; Rehman, Y.; Sheik, C.S.; Abbas, Z.; Rehman, S. Complete genome analysis of a Siphoviridae phage TSK1 showing biofilm removal potential against Klebsiella pneumoniae. Sci. Rep. 2018, 8, 17904. [Google Scholar] [CrossRef]
- Verma, V.; Harjai, K.; Chhibber, S. Characterization of a T7-Like Lytic bacteriophage of Klebsiella pneumoniae B5055: A potential therapeutic agent. Curr. Microbiol. 2009, 59, 274–281. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.R.; Axler, R.P.; Hicks, R.E. Effects of freezing and storage temperature on MS2 viability. J. Virol. Methods 2004, 122, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Jończyk, E.; Kłak, M.; Międzybrodzki, R.; Górski, A. The influence of external factors on bacteriophages—Review. Folia Microbiol. 2011, 56, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Jepson, C.D.; March, J.B. Bacteriophage lambda is a highly stable DNA vaccine delivery vehicle. Vaccine 2004, 22, 2413–2419. [Google Scholar] [CrossRef] [PubMed]
- Philipson, C.W.; Voegtly, L.J.; Lueder, M.R.; Long, K.A.; Rice, G.K.; Frey, K.G.; Biswas, B.; Cer, R.Z.; Hamilton, T.; Bishop-Lilly, K.A. Characterizing phage genomes for therapeutic applications. Viruses 2018, 10, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Kumar, D.; Poluri, K.M. Elucidating the pH-dependent structural transition of T7 bacteriophage endolysin. Biochemistry 2016, 55, 4614–4625. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.C.; Schmelcher, M.; Rodriguez-Rubio, L.; Klumpp, J.; Pritchard, D.G.; Dong, S.; Donovan, D.M. Endolysins as antimicrobials. In Advances in Virus Research 2012, 83, 299–365. [Google Scholar]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moak, M.; Molineux, I.J. Peptidoglycan hydrolytic activities associated with bacteriophage virions. Mol. Microbiol. 2004, 51, 1169–1183. [Google Scholar] [CrossRef]
- Shi, Y.; Yan, Y.; Ji, W.; Du, B.; Meng, X.; Wang, H.; Sun, J. Characterization and determination of holin protein of Streptococcus suis bacteriophage SMP in heterologous host. Virol. J. 2012, 9. [Google Scholar] [CrossRef] [Green Version]
- Tu, J.; Park, T.; Morado, D.R.; Hughes, K.T.; Molineux, I.J.; Liu, J. Dual host specificity of phage SP6 is facilitated by tailspike rotation. Virology 2017, 507, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Steven, A.C.; Trus, B.L.; Maizel, J.V.; Unser, M.; Parry, D.A.D.; Wall, J.S.; Hainfeld, J.F.; Studier, F.W. Molecular substructure of a viral receptor-recognition protein. J. Mol. Biol. 1988, 200, 351–365. [Google Scholar] [CrossRef]
- Studier, F.W. Bacteriophage T7. Science 1972, 176, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Doval, C.; van Raaij, M.J. Structure of the receptor-binding carboxy-terminal domain of bacteriophage T7 tail fibers. Proc. Natl. Acad. Sci. USA 2012, 109, 9390–9395. [Google Scholar] [CrossRef] [Green Version]
- Manohar, P.; Tamhankar, A.J.; Lundborg, C.S.; Nachimuthu, R. Therapeutic characterization and efficacy of bacteriophage cocktails infecting Escherichia coli, Klebsiella pneumoniae, and Enterobacter species. Front. Microbiol. 2019, 10, 574. [Google Scholar] [CrossRef] [Green Version]
- Pyra, A.; Urbańska, N.; Filik, K.; Tyrlik, K.; Brzozowska, E. Biochemical features of the novel Tail Tubular Protein A of Yersinia phage phiYeO3-12. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Cuervo, A.; Pulido-Cid, M.; Chagoyen, M.; Arranz, R.; González-García, V.A.; Garcia-Doval, C.; Castón, J.R.; Valpuesta, J.M.; van Raaij, M.J.; Martín-Benito, J.; et al. Structural characterization of the bacteriophage T7 tail machinery. J. Biol. Chem. 2013, 288, 26290–26299. [Google Scholar] [CrossRef] [Green Version]
- Pyra, A.; Brzozowska, E.; Pawlik, K.; Gamian, A.; Dauter, M.; Dauter, Z. Tail tubular protein A: A dual-function tail protein of Klebsiella pneumoniae bacteriophage KP32. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassa, T.; Chhibber, S. Thermal treatment of the bacteriophage lysate of Klebsiella pneumoniae B5055 as a step for the purification of capsular depolymerase enzyme. J. Virol. Methods 2012, 179, 135–141. [Google Scholar] [CrossRef]
- Donlan, R.M. Preventing biofilms of clinically relevant organisms using bacteriophage. Trends Microbiol. 2009, 17, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Chen, Y.; Yang, Z.; Zhang, Y.; You, B.; Liu, X.; Chen, P.; Liu, M.; Zhang, C.; Luo, X.; et al. Characterization and genome sequencing of a novel T7-like lytic phage, kpssk3, infecting carbapenem-resistant Klebsiella pneumoniae. Arch. Virol. 2020, 165, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Buttimer, C.; Lucid, A.; Neve, H.; Franz, C.; O’Mahony, J.; Turner, D.; Lavigne, R.; Coffey, A. Pectobacterium atrosepticum phage vB_PatP_CB5: A member of the proposed genus ‘Phimunavirus’. Viruses 2018, 10, 394. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.; Tavares, P.; Petit, M.-A.; Guérois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genom. 2014, 15, 1027. [Google Scholar] [CrossRef] [Green Version]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and their genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, V.B.; Feiss, M. Mechanisms of DNA packaging by large double-stranded DNA viruses. Annu. Rev. Virol. 2015, 2, 351–378. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Rao, V.B.; Rossmann, M.G. Genome packaging in viruses. Curr. Opin. Struct. Biol. 2010, 20, 114–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanamaru, S.; Kondabagil, K.; Rossmann, M.G.; Rao, V.B. The functional domains of bacteriophage T4 terminase. J. Biol. Chem. 2004, 279, 40795–40801. [Google Scholar] [CrossRef] [Green Version]
- Teng, T.; Li, Q.; Liu, Z.; Li, X.; Liu, Z.; Liu, H.; Liu, F.; Xie, L.; Wang, H.; Zhang, L.; et al. Characterization and genome analysis of novel Klebsiella phage Henu1 with lytic activity against clinical strains of Klebsiella pneumoniae. Arch. Virol. 2019, 164, 2389–2393. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.H.; Bowman, C.A.; Russell, D.A.; Jacobs-Sera, D.; Asai, D.J.; Cresawn, S.G.; Jacobs, W.R.; Hendrix, R.W.; Lawrence, J.G.; Hatfull, G.F. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. eLife 2015, 4, e06416. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, L.; Lins, B.; Barrett, J.; Montgomery, A.; Trapani, S.; Schindler, A.; Christie, G.E.; Cresawn, S.G.; Temple, L. Genomic characterization of six novel Bacillus pumilus bacteriophages. Virology 2013, 444, 374–383. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Source | KPP-5 Phage Isolate | |
|---|---|---|---|
| ST | EOP | ||
| K. pneumoniae-1 (host strain) | food | + | H |
| K. pneumoniae-2 | food | + | M |
| K. pneumoniae-3 | food | + | M |
| K. pneumoniae-4 | food | + | H |
| K. pneumoniae-5 | food | + | H |
| K. pneumoniae-6 | clinical | + | H |
| K. pneumoniae-7 | clinical | + | H |
| K. pneumoniae-8 | clinical | + | M |
| K. pneumoniae-9 | clinical | + | H |
| K. pneumoniae-10 | clinical | + | M |
| K. pneumoniae-11 | clinical | + | H |
| K. pneumoniae-12 | clinical | + | H |
| K. pneumoniae-13 | clinical | + | H |
| K. pneumoniae-14 | clinical | + | H |
| K. pneumoniae-15 | clinical | + | H |
| K. pneumoniae-16 | clinical | + | H |
| K. pneumoniae-17 | clinical | + | M |
| K. pneumoniae-18 | clinical | + | H |
| K. pneumoniae-19 | clinical | + | H |
| E. coli-1 | clinical | − | N |
| E. coli-2 | clinical | − | N |
| E. coli-3 | clinical | − | N |
| E. coli-4 | clinical | − | N |
| E. coli-5 | clinical | − | N |
| E. coli-6 | clinical | − | N |
| S. Typhimurium-1 | food | − | N |
| S. Typhimurium-2 | food | − | N |
| S. Typhimurium-3 | food | − | N |
| P. aeruginosa-1 | clinical | − | N |
| P. aeruginosa-2 | clinical | − | N |
| P. aeruginosa-3 | food | − | N |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sofy, A.R.; El-Dougdoug, N.K.; Refaey, E.E.; Dawoud, R.A.; Hmed, A.A. Characterization and Full Genome Sequence of Novel KPP-5 Lytic Phage against Klebsiella pneumoniae Responsible for Recalcitrant Infection. Biomedicines 2021, 9, 342. https://doi.org/10.3390/biomedicines9040342
Sofy AR, El-Dougdoug NK, Refaey EE, Dawoud RA, Hmed AA. Characterization and Full Genome Sequence of Novel KPP-5 Lytic Phage against Klebsiella pneumoniae Responsible for Recalcitrant Infection. Biomedicines. 2021; 9(4):342. https://doi.org/10.3390/biomedicines9040342
Chicago/Turabian StyleSofy, Ahmed R., Noha K. El-Dougdoug, Ehab E. Refaey, Rehab A. Dawoud, and Ahmed A. Hmed. 2021. "Characterization and Full Genome Sequence of Novel KPP-5 Lytic Phage against Klebsiella pneumoniae Responsible for Recalcitrant Infection" Biomedicines 9, no. 4: 342. https://doi.org/10.3390/biomedicines9040342