
SSCS: A Stage Supervised Subtyping System for Colorectal Cancer


Abstract
:1. Introduction
2. Methods
2.1. CRC Cohort Datasets
2.2. Gene Set Enrichment Analysis (GSEA)
2.3. Survival and Cox PH Regression Analysis
2.4. Unsupervised Clustering and Random Forest Classifiers
2.5. Statistical Analyses
3. Results
3.1. Evaluations of Three Previous CRC Subtyping Systems
3.2. TNM Stage Remains an Influential Factor for CRC Subtype Analysis
3.3. Identification of Five Stage Supervised CRC Subtypes
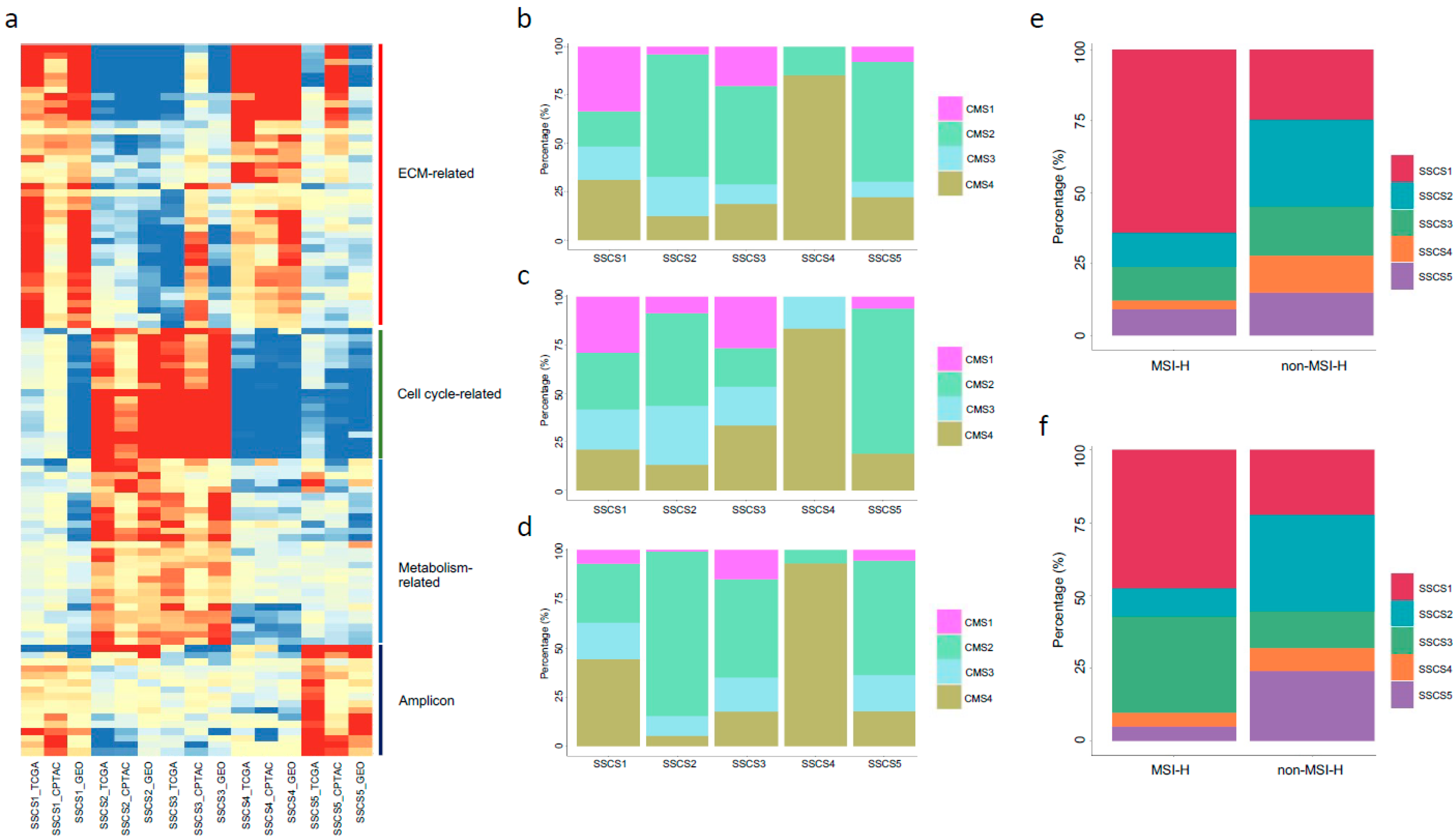
3.4. Molecular Features of the Five Subtypes
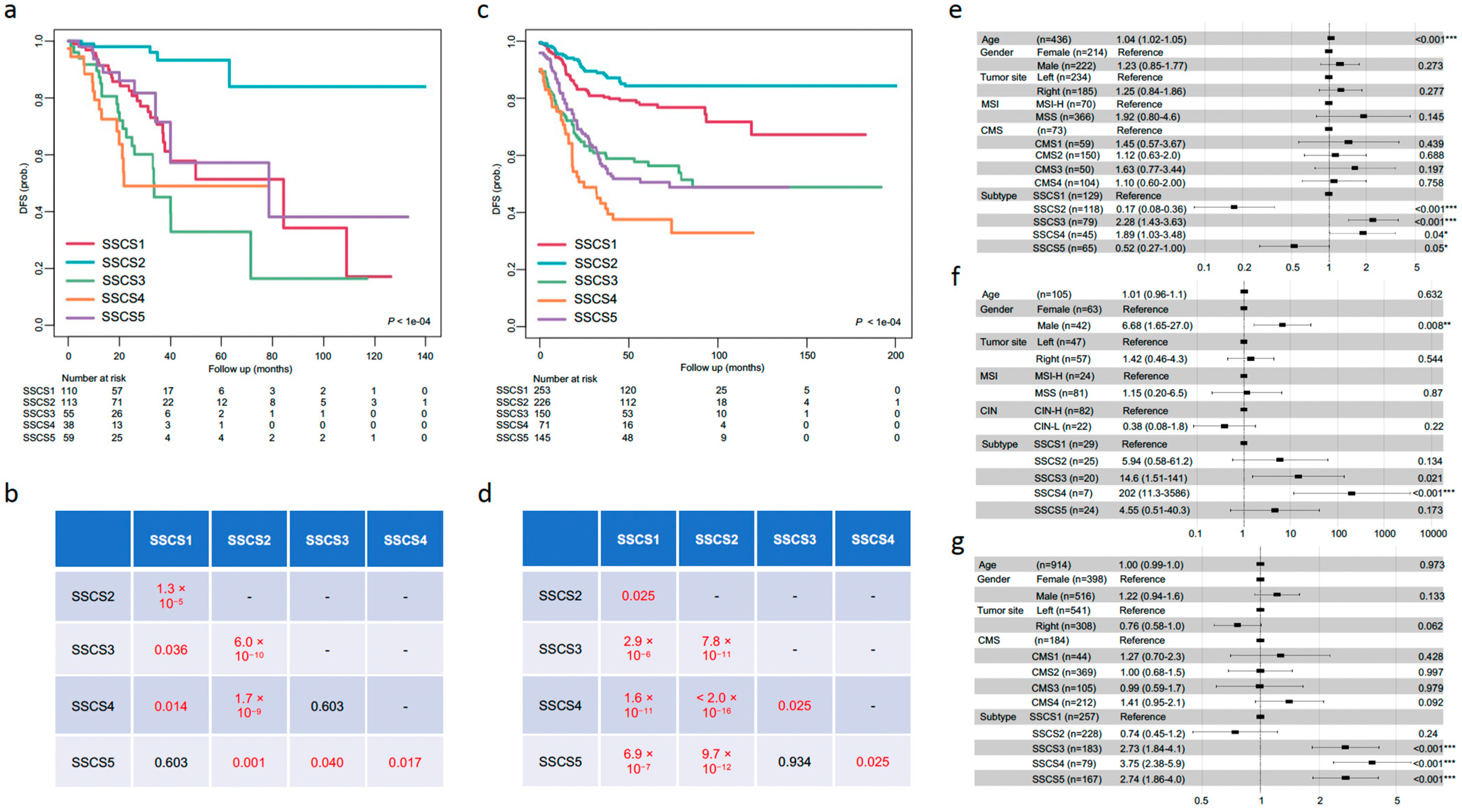
3.5. Prognostic Value of SSCS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nguyen, H.T.; Duong, H.-Q. The Molecular Characteristics of Colorectal Cancer: Implications for Diagnosis and Therapy. Oncol. Lett. 2018, 16, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, J.H.; Kim, J.H.; Lee, J.; Kye, B.-H.; Lee, S.C.; Lee, I.K.; Kang, W.K.; Cho, H.-M.; Lee, Y.S. Addition of V-Stage to Conventional TNM Staging to Create the TNVM Staging System for Accurate Prediction of Prognosis in Colon Cancer: A Multi-Institutional Retrospective Cohort Study. Biomedicines 2021, 9, 888. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic Instabilities in Human Cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Goel, A. Molecular Classification and Correlates in Colorectal Cancer. J. Mol. Diagn. 2008, 10, 13–27. [Google Scholar] [CrossRef] [Green Version]
- Vilar, E.; Gruber, S.B. Microsatellite Instability in Colorectal Cancer-the Stable Evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Tóth, G.; Gáspári, Z.; Jurka, J. Microsatellites in Different Eukaryotic Genomes: Survey and Analysis. Genome Res. 2000, 10, 967–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, H.; Zaanan, A.; Sinicrope, F.A. Microsatellite Instability Testing and Its Role in the Management of Colorectal Cancer. Curr. Treat. Options Oncol. 2015, 16, 30. [Google Scholar] [CrossRef]
- Yamamoto, H.; Imai, K. Microsatellite Instability: An Update. Arch. Toxicol. 2015, 89, 899–921. [Google Scholar] [CrossRef]
- Nojadeh, J.N.; Behrouz Sharif, S.; Sakhinia, E. Microsatellite Instability in Colorectal Cancer. EXCLI J. 2018, 17, 159–168. [Google Scholar]
- Kim, G.P.; Colangelo, L.H.; Samuel Wieand, H.; Paik, S.; Kirsch, I.R.; Wolmark, N.; Allegra, C.J. Prognostic and Predictive Roles of High-Degree Microsatellite Instability in Colon Cancer: A National Cancer Institute–National Surgical Adjuvant Breast and Bowel Project Collaborative Study. J. Clin. Oncol. 2007, 25, 767–772. [Google Scholar] [CrossRef]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite Instability: A Review of What the Oncologist Should Know. Cancer Cell Int. 2020, 20, 16. [Google Scholar] [CrossRef] [Green Version]
- Budinska, E.; Popovici, V.; Tejpar, S.; D’Ario, G.; Lapique, N.; Sikora, K.O.; Di Narzo, A.F.; Yan, P.; Hodgson, J.G.; Weinrich, S.; et al. Gene Expression Patterns Unveil a New Level of Molecular Heterogeneity in Colorectal Cancer. J. Pathol. 2013, 231, 63–76. [Google Scholar] [CrossRef]
- Marisa, L.; de Reyniès, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.-C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef] [Green Version]
- Roepman, P.; Schlicker, A.; Tabernero, J.; Majewski, I.; Tian, S.; Moreno, V.; Snel, M.H.; Chresta, C.M.; Rosenberg, R.; Nitsche, U.; et al. Colorectal Cancer Intrinsic Subtypes Predict Chemotherapy Benefit, Deficient Mismatch Repair and Epithelial-to-Mesenchymal Transition. Int. J. Cancer 2014, 134, 552–562. [Google Scholar] [CrossRef]
- Felipe De Sousa, E.M.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; de Rooij, L.P.; de Jong, J.H.; de Boer, O.J.; van Leersum, R.; Bijlsma, M.F.; et al. Poor-Prognosis Colon Cancer Is Defined by a Molecularly Distinct Subtype and Develops from Serrated Precursor Lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.G.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A Colorectal Cancer Classification System That Associates Cellular Phenotype and Responses to Therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [Green Version]
- Schlicker, A.; Beran, G.; Chresta, C.M.; McWalter, G.; Pritchard, A.; Weston, S.; Runswick, S.; Davenport, S.; Heathcote, K.; Castro, D.A.; et al. Subtypes of Primary Colorectal Tumors Correlate with Response to Targeted Treatment in Colorectal Cell Lines. BMC Med. Genom. 2012, 5, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Mooi, J.K.; Wirapati, P.; Asher, R.; Lee, C.K.; Savas, P.; Price, T.J.; Townsend, A.; Hardingham, J.; Buchanan, D.; Williams, D.; et al. The Prognostic Impact of Consensus Molecular Subtypes (CMS) and Its Predictive Effects for Bevacizumab Benefit in Metastatic Colorectal Cancer: Molecular Analysis of the AGITG MAX Clinical Trial. Ann. Oncol. 2018, 29, 2240–2246. [Google Scholar] [CrossRef] [PubMed]
- Lenz, H.-J.; Ou, F.-S.; Venook, A.P.; Hochster, H.S.; Niedzwiecki, D.; Goldberg, R.M.; Mayer, R.J.; Bertagnolli, M.M.; Blanke, C.D.; Zemla, T.; et al. Impact of Consensus Molecular Subtype on Survival in Patients with Metastatic Colorectal Cancer: Results from CALGB/SWOG 80405 (alliance). J. Clin. Oncol. 2019, 37, 1876–1885. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Qvortrup, C. Does the Consensus Molecular Subtypes Classification Add to Selection of Precision Medicine in Patients with Metastatic Colorectal Cancer? Dig. Med. Res. 2020, 3. [Google Scholar] [CrossRef]
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177, 1035–1049.e19. [Google Scholar] [CrossRef] [Green Version]
- Mestres, A.C.; Llergo, A.B.; Attolini, C.S.-O.; Mestres, M.A.C. Package “Mcsurvdata.”. Available online: https://bioconductor.org/packages/3.14/data/experiment/ (accessed on 15 August 2019).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Väremo, L.; Nielsen, J.; Nookaew, I. Enriching the Gene Set Analysis of Genome-Wide Data by Incorporating Directionality of Gene Expression and Combining Statistical Hypotheses and Methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar] [CrossRef]
- Kolde, R. Pheatmap: Pretty Heatmaps. R Package Version 2012, 1. Available online: https://rdrr.io/cran/pheatmap/ (accessed on 15 August 2019).
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Survminer: Drawing Survival Curves using’ggplot2′. R Package Version 0.3. 2017, 1. Available online: https://rpkgs.datanovia.com/survminer/ (accessed on 15 August 2019).
- Cirocchi, R.; Cesare Campanile, F.; Di Saverio, S.; Popivanov, G.; Carlini, L.; Pironi, D.; Tabola, R.; Vettoretto, N. Laparoscopic versus Open Colectomy for Obstructing Right Colon Cancer: A Systematic Review and Meta-Analysis. J. Visc. Surg. 2017, 154, 387–399. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A Class Discovery Tool with Confidence Assessments and Item Tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [Green Version]
- Liaw, A.; Wiener, M. Others Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally Portraying the Tissue Cellular Heterogeneity Landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Yoo, B.B.; Mazmanian, S.K. The Enteric Network: Interactions between the Immune and Nervous Systems of the Gut. Immunity 2017, 46, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Hirota, T.; Ohashi, Y.; Kodaira, S. Prospective Trial of Adjuvant Chemotherapy for Colon Cancer Study Group (PAC) Significance of Histologic Type of Primary Lesion and Metastatic Lymph Nodes as a Prognostic Factor in Stage III Colon Cancer. Dis. Colon Rectum 2006, 49, 982–992. [Google Scholar] [CrossRef]
- Compton, C.C. Optimal Pathologic Staging: Defining Stage II Disease. Clin. Cancer Res. 2007, 13, 6862s–6870s. [Google Scholar] [CrossRef] [Green Version]
- Rohaan, M.W.; van den Berg, J.H.; Kvistborg, P.; Haanen, J.B.A.G. Adoptive Transfer of Tumor-Infiltrating Lymphocytes in Melanoma: A Viable Treatment Option. J. Immunother Cancer 2018, 6, 102. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Xu, F.; Zhong, K.; Wang, S.; Huang, L.; Chen, W. Activated Tumor-Infiltrating Fibroblasts Predict Worse Prognosis in Breast Cancer Patients. J. Cancer 2018, 9, 3736–3742. [Google Scholar] [CrossRef] [Green Version]
- Idos, G.E.; Kwok, J.; Bonthala, N.; Kysh, L.; Gruber, S.B.; Qu, C. The Prognostic Implications of Tumor Infiltrating Lymphocytes in Colorectal Cancer: A Systematic Review and Meta-Analysis. Sci. Rep. 2020, 10, 3360. [Google Scholar] [CrossRef]
- Sichien, D.; Lambrecht, B.N.; Guilliams, M.; Scott, C.L. Development of Conventional Dendritic Cells: From Common Bone Marrow Progenitors to Multiple Subsets in Peripheral Tissues. Mucosal Immunol. 2017, 10, 831–844. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Label | Character | Classification | Disadvantage | TCGA | CPTAC | GEO |
|---|---|---|---|---|---|---|---|
| Stage | Early | Better prognosis | Supervised | No molecular basis | 247 (56.7%) | 54 (51.4%) | 485 (53.1%) |
| Advanced | ECM and cell proliferation pathways up | 189 (43.3%) | 51 (48.6%) | 429 (46.9%) | |||
| MSI | MSI-H | Immune pathways up | Supervised | High heterogeneity within the non-MSI-H group | 70 (16.0%) | 24 (22.8%) | 0 (0) |
| Non-MSI-H | Significant proportion of patients | 366 (84.0%) | 81 (77.2%) | 914 (100.0%) | |||
| CMS | CMS1 | MSI-H | Unsupervised | Has an unassigned group | 59 (13.5%) | 14 (13.3%) | 44 (4.8%) |
| CMS2 | Canonical cancer pathways up | 150 (34.4%) | 33 (31.4%) | 369 (40.4%) | |||
| CMS3 | Metabolic | 50 (11.5%) | 16 (15.3%) | 105 (11.5%) | |||
| CMS4 | EMT | 104 (23.9%) | 21 (20.0%) | 212 (23.2%) | |||
| NOLBL | Unassigned | 73 (16.7%) | 21 (20.0%) | 184 (20.1%) | |||
| Stage + MSI | Early_MSI-H | More MSI-H patients | Supervised | Non-distinction between early- and advanced-cases in MSI-H patients | 53 (12.1%) | 14 (13.3%) | - |
| Early_non-MSI-H | Oxidation pathway up | 194 (44.5%) | 40 (38.1%) | - | |||
| Advanced_MSI-H | Fewer patients | 17 (3.9%) | 10 (9.5%) | - | |||
| Advanced_non-MSI-H | Worst prognosis | 172 (39.5%) | 41 (39.1%) | - | |||
| Stage + CMS | CMS1-4 (in early-stage) | Distinctive | Supervised | CMS with poor performances in advanced case | 214 (59.0%) | 47 (56.0%) | 405 (55.5%) |
| CMS1-4 (in advanced-stage) | Non-distinctive | 149 (41.0%) | 37 (44.0%) | 325 (44.5%) | |||
| SSCS | SSCS1 | MSI-H | Supervised + unsupervised | No tumor grade information | 129 (29.6%) | 29 (27.6%) | 257 (28.1%) |
| SSCS2 | ECM pathway down; better prognosis | 118 (27.1%) | 25 (23.8%) | 228 (25.0%) | |||
| SSCS3 | Cell cycle pathway up; cDC infiltration | 79 (18.1%) | 20 (19.1%) | 183 (20.0%) | |||
| SSCS4 | Fibroblast infiltration; worst prognosis | 45 (10.3%) | 7 (6.7%) | 79 (8.6%) | |||
| SSCS5 | Amplicon pathway up | 65 (14.9%) | 24 (22.8%) | 167 (18.3%) |
| Subtype | Neuron | Fibroblast | Memory CD4+ T | cDC | Naive B | Naive CD4+ T | |
|---|---|---|---|---|---|---|---|
| DFS | SSCS1 | 0.062 | 0.500 | 0.980 | 0.320 | 0.680 | 1 |
| SSCS2 | 0.080 | 1 | 0.130 | 0.330 | 0.130 | 1 | |
| SSCS3 | 0.200 | 1 | 0.059 | 0.008 ** | 0.830 | 1 | |
| SSCS4 | 0.290 | 0.001 ** | 0.540 | 0.570 | 0.870 | 1 | |
| SSCS5 | 0.710 | 1 | 0.320 | 0.520 | 1 | 0.540 | |
| All | 0.064 | 0.120 | 0.090 | 0.008 ** | 0.140 | 0.460 | |
| OS | SSCS1 | 0.710 | 0.190 | 0.510 | 0.810 | 0.750 | 1 |
| SSCS2 | 0.260 | 1 | 0.086 | 0.630 | 0.390 | 1 | |
| SSCS3 | 0.170 | 1 | 0.190 | 0.058 | 0.450 | 0.870 | |
| SSCS4 | 0.270 | 0.340 | 0.120 | 0.560 | 0.310 | 0.620 | |
| SSCS5 | 0.580 | 0.200 | 0.058 | 0.570 | 1 | 1 | |
| All | 0.100 | 0.210 | 0.610 | 0.510 | 0.810 | 0.720 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Pan, Y. SSCS: A Stage Supervised Subtyping System for Colorectal Cancer. Biomedicines 2021, 9, 1815. https://doi.org/10.3390/biomedicines9121815
Zhao L, Pan Y. SSCS: A Stage Supervised Subtyping System for Colorectal Cancer. Biomedicines. 2021; 9(12):1815. https://doi.org/10.3390/biomedicines9121815
Chicago/Turabian StyleZhao, Lan, and Yi Pan. 2021. "SSCS: A Stage Supervised Subtyping System for Colorectal Cancer" Biomedicines 9, no. 12: 1815. https://doi.org/10.3390/biomedicines9121815