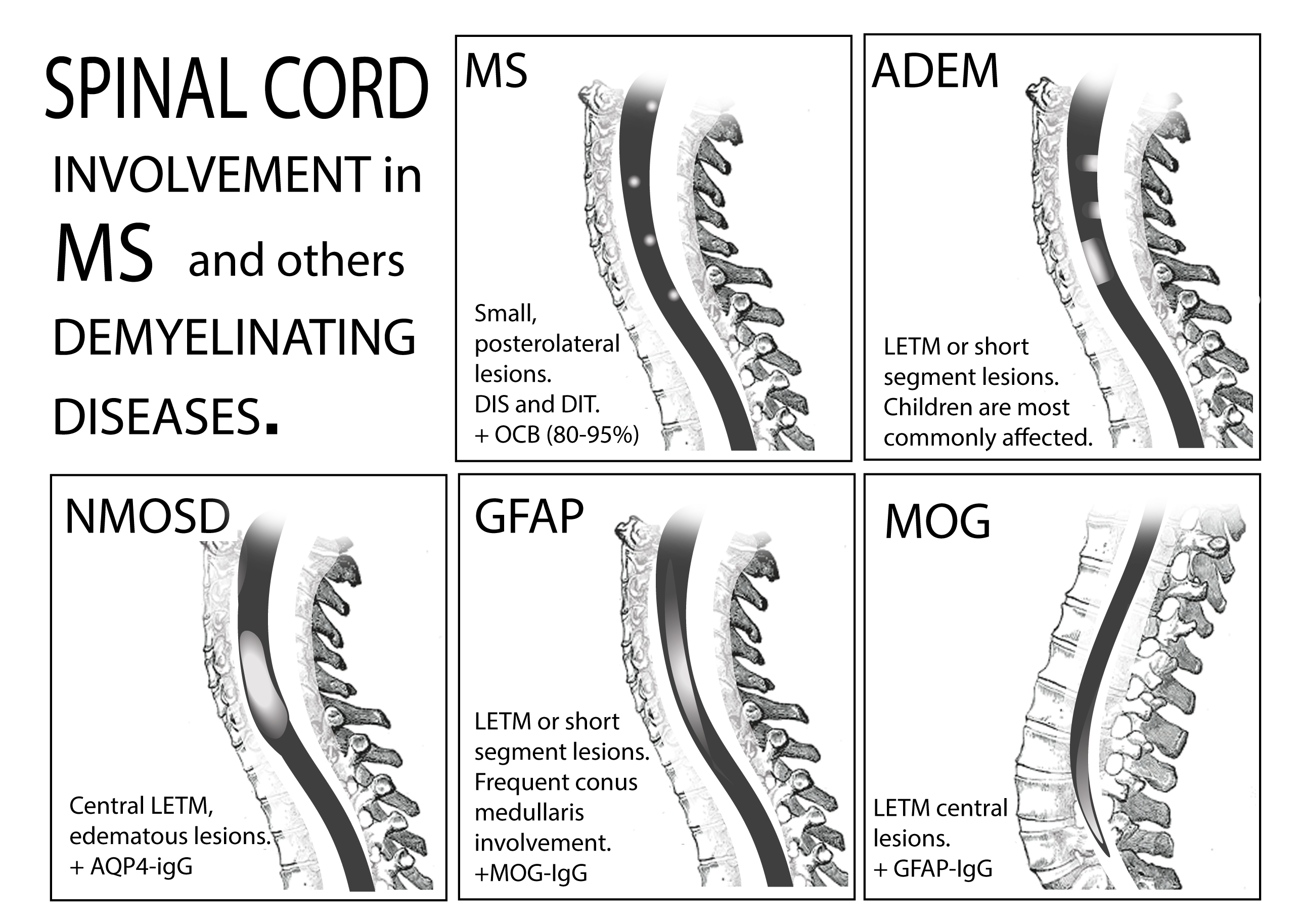
Spinal Cord Involvement in MS and Other Demyelinating Diseases


Abstract
:
1. Introduction
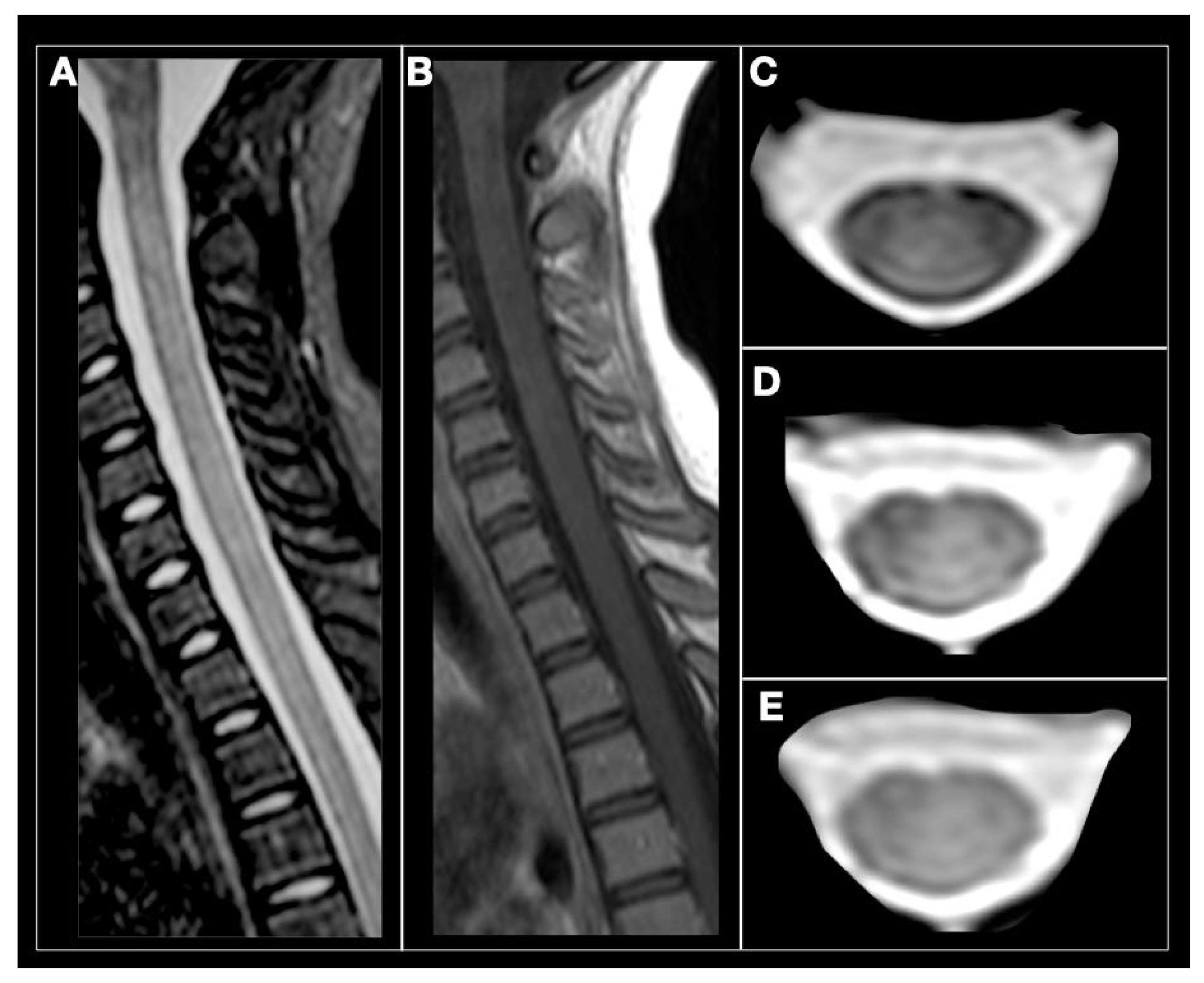
2. Multiple Sclerosis
3. Acute Disseminated Encephalomyelitis
4. Neuromyelitis Optica Spectrum-Disorder
5. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease
6. Glial Fibrillary Acid Protein Antibody-Associated Myelitis
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barreras, P.; Fitzgerald, K.C.; Mealy, M.A.; Jimenez, J.A.; Becker, D.; Newsome, S.D.; Levy, M.; Gailloud, P.; Pardo, C.A. Clinical biomarkers differentiate myelitis from vascular and other causes of myelopathy. Neurology 2018, 90, E12–E21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Transverse Myelitis Consortium Working Group. Proposed diagnostic criteria and nosology of acute transverse myelitis. Neurology 2002, 59, 499–505. [Google Scholar] [CrossRef] [PubMed]
- de Seze, J.; Lanctin, C.; Lebrun, C.; Malikova, I.; Papeix, C.; Wiertlewski, S.; Pelletier, J.; Gout, O.; Clerc, C.; Moreau, C.; et al. Idiopathic acute transverse myelitis: Application of the recent diagnostic criteria. Neurology 2005, 65, 1950–1953. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Weinshenker, B.G. An approach to the diagnosis of acute transverse myelitis. Semin. Neurol. 2008, 28, 105–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GBD 2015 Neurological Disorders Collaborator Group. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef] [Green Version]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- McFarland, H.F.; Martin, R. Multiple sclerosis: A complicated picture of autoimmunity. Nat. Immunol. 2007, 8, 913–919. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Compston, A.; Mc Donald, I.; Noseworthy, J.; Lassmann, H.; Miller, D.; Smith, K.; Wekerle, H.; Confavreux, C. McAlpine’s Multiple Sclerosis, 4th ed.; Churchill-Livingstone: London, UK, 2005. [Google Scholar]
- Cordonnier, C.; De Seze, J.; Breteau, G.; Ferriby, D.; Michelin, E.; Stojkovic, T.; Pruvo, J.P.; Vermersch, P. Prospective study of patients presenting with acute partial transverse myelopathy. J. Neurol. 2003, 250, 1447–1452. [Google Scholar] [CrossRef]
- Kantarci, O.H. Phases and Phenotypes of Multiple Sclerosis. Continuum (Minneap Minn) 2019, 25, 636–654. [Google Scholar] [CrossRef]
- Ingle, G.T.; Sastre-Garriga, J.; Miller, D.H.; Thomson, A.J. Is inflammation important in early PPMS? A longitudinal MRI study. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1255–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, M.W.; Greenfield, J.; Javizian, O.; Deighton, S.; Wall, W.; Metz, L.M. The natural history of early versus late disability accumulation in primary progressive M.S. J. Neurol. Neurosurg. Psychiatry 2015, 86, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Vukusic, S.; Confavreux, C. Primary and secondary progressive multiple sclerosis. J. Neurol. Sci. 2003, 206, 153–155. [Google Scholar] [CrossRef]
- Marrodan, M.; Bensi, C.; Pappolla, A.; Rojas, J.I.; Gaitán, M.I.; Ysrraelit, M.C.; Negrotto, L.; Fiol, M.P.; Patrucco, L.; Cristiano, E. Disease activity impacts disability progression in primary progressive multiple sclerosis. Mult. Scler. Relat. Disord. 2020, 39, 101892. [Google Scholar] [CrossRef] [PubMed]
- Androdias, G.; Reynolds, R.; Chanal, M.; Ritleng, C.; Confavreux, C.; Nataf, S. Meningeal T cells associate with diffuse axonal loss in multiple sclerosis spinal cords. Ann. Neurol. 2010, 68, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.H.; McCarron, R.; McFarlin, D.E.; Raine, C.S. Adoptively transferred acute and chronic relapsing autoimmune encephalomyelitis in the PL/J mouse and observations on altered pathology by intercurrent virus infection. Lab. Investig. 1987, 57, 499–512. [Google Scholar]
- Schwartz, M.; Butovsky, O.; Brück, W.; Hanisch, U.K. Microglial phenotype: Is the commitment reversible? Trends Neurosci. 2006, 29, 68–74. [Google Scholar] [CrossRef]
- Krieger, S.C.; Lublin, F.D. Location, location, location. Mult. Scler. J. 2018, 24, 1396–1398. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Lycklama, G.; Thompson, A.; Filippi, M.; Miller, D.; Polman, C.; Fazekas, F.; Barkhof, F. Spinal-cord MRI in multiple sclerosis. Lancet Neurol. 2003, 2, 555–562. [Google Scholar] [CrossRef]
- Lycklama, À.; Nijeholt, G.J.; Castelijns, J.A.; Weerts, J.; Adèr, H.; van Waesberghe, J.H.; Polman, C.; Barkhof, F. Sagittal MR of multiple sclerosis in the spinal cord: Fast versus conventional spin-echo imaging. Am. J. Neuroradiol. 1998, 19, 355–360. [Google Scholar]
- Gass, A.; Rocca, M.A.; Agosta, F.; Ciccarelli, O.; Chard, D.; Valsasina, P.; Brooks, J.C.; Bischof, A.; Eisele, P.; Kappos, L.; et al. MRI monitoring of pathological changes in the spinal cord in patients with multiple sclerosis. Lancet Neurol. 2015, 14, 443–454. [Google Scholar] [CrossRef]
- Rovira, Á.; Wattjes, M.P.; Tintoré, M.; Tur, C.; Yousry, T.A.; Sormani, M.P.; De Stefano, N.; Filippi, M.; Auger, C.; Rocca, M.A.; et al. Evidence-based guidelines: MAGNIMS consensus guidelines on the use of MRI in multiple sclerosis—Clinical implementation in the diagnostic process. Nat. Rev. Neurol. 2015, 11, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Bergers, E.; Bot, J.C.J.; De Groot, C.J.A.; Polman, C.H.; Lycklama, G.J.; Nijeholt, Á.; Castelijns, J.A.; van der Valk, P.; Barkhof, F. Axonal damage in the spinal cord of MS patients occurs largely independent of T2 MRI lesions. Neurology 2002, 59, 1766–1771. [Google Scholar] [CrossRef]
- Weier, K.; Mazraeh, J.; Naegelin, Y.; Thoeni, A.; Hirsch, J.G.; Fabbro, T.; Bruni, N.; Duyar, H.; Bendfeldt, K.; Radue, E.W. Biplanar MRI for the assessment of the spinal cord in multiple sclerosis. Mult. Scler. J. 2012, 18, 1560–1569. [Google Scholar] [CrossRef] [PubMed]
- Breckwoldt, M.O.; Gradl, J.; Hähnel, S.; Hielscher, T.; Wildemann, B.; Diem, R.; Platten, M.; Wick, W.; Heiland, S.; Bendszus, M. Increasing the sensitivity of MRI for the detection of multiple sclerosis lesions by long axial coverage of the spinal cord: A prospective study in 119 patients. J. Neurol. 2017, 264, 341–349. [Google Scholar] [CrossRef]
- Martin, N.; Malfair, D.; Zhao, Y.; Li, D.; Traboulsee, A.; Lang, F.; Vertinsky, A.T. Comparison of MERGE and axial T2-weighted fast spin-echo sequences for detection of multiple sclerosis lesions in the cervical spinal cord. Am. J. Roentgenol. 2012, 199, 157–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalewski, N.L.; Morris, P.P.; Weinshenker, B.G.; Lucchinetti, C.F.; Guo, Y.; Pittock, S.J.; Krecke, K.N.; Kaufmann, T.J.; Wingerchuk, D.M.; Kumar, N.; et al. Ring-enhancing spinal cord lesions in neuromyelitis optica spectrum disorders. J. Neurol. Neurosurg. Psychiatry 2017, 88, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, O.; Cohen, J.A.; Reingold, S.C.; Weinshenker, B.G. Spinal cord involvement in multiple sclerosis and neuromyelitis optica spectrum disorders. Lancet Neurol. 2019, 18, 185–197. [Google Scholar] [CrossRef]
- de Seze, J. Acute myelopathies: Clinical, laboratory and outcome profiles in 79 cases. Brain 2001, 124, 1509–1521. [Google Scholar] [CrossRef]
- Kitley, J.L.; Leite, M.I.; George, J.S.; Palace, J.A. The differential diagnosis of longitudinally extensive transverse myelitis. Mult. Scler. J. 2012, 18, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, R.; Ciccarelli, O.; Barkhof, F.; De Stefano, N.; Enzinger, C.; Filippi, M.; Hofer, M.; Paul, F.; Preziosa, P.; Rovira, A.; et al. The current role of MRI in differentiating multiple sclerosis from its imaging mimics. Nat. Rev. Neurol. 2018, 14, 199–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarci, O.H.; Lebrun, C.; Siva, A.; Keegan, M.B.; Azevedo, C.J.; Inglese, M.; Tintoré, M.; Newton, B.D.; Durand-Dubief, F.; Amato, M.P.; et al. Primary Progressive Multiple Sclerosis Evolving from Radiologically Isolated Syndrome. Ann. Neurol. 2016, 79, 288–294. [Google Scholar] [CrossRef]
- O’Riordan, J.I.; Thompson, A.J.; Kingsley, D.P.E.; MacManus, D.G.; Kendall, B.E.; Rudge, P.; McDonald, W.I.; Miller, D.H. The prognostic value of brain MRI in clinically isolated syndromes of the CNS. A 10-year follow-up. Brain 1998, 121, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bot, J.C.J.; Barkhof, F.; Polman, C.H.; Lycklama, Á.; Nijeholt, G.J.; de Groot, V.; Bergers, E.; Ader, H.J.; Castelijns, J.A. Spinal cord abnormalities in recently diagnosed MS patients: Added value of spinal MRI examination. Neurology 2004, 62, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Arrambide, G.; Tintore, M.; Auger, C.; Río, J.; Castilló, J.; Vidal-Jordana, A.; Galán, I.; Nos, C.; Comabella, M.; Mitjana, R.; et al. Lesion topographies in multiple sclerosis diagnosis: A reappraisal. Neurology 2017, 89, 2351–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, W.J.; Altmann, D.R.; Alves Da Mota, P.; Swanton, J.K.; Miszkiel, K.A.; Wheeler-Kingshott, C.G.; Ciccarelli, O.; Miller, D.H. Association of asymptomatic spinal cord lesions and atrophy with disability 5 years after a clinically isolated syndrome. Mult. Scler. 2017, 23, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Zecca, C.; Disanto, G.; Sormani, M.P.; Riccitelli, G.C.; Cianfoni, A.; Del Grande, F.; Pravatà, E.; Gobbi, C. Relevance of asymptomatic spinal MRI lesions in patients with multiple sclerosis. Mult. Scler. 2016, 22, 782–791. [Google Scholar] [CrossRef]
- Vukusic, S.; Confavreux, C. Prognostic factors for progression of disability in the secondary progressive phase of multiple sclerosis. J. Neurol. Sci. 2003, 206, 135–137. [Google Scholar] [CrossRef]
- Biberacher, V.; Boucard, C.C.; Schmidt, P.; Engl, C.; Buck, D.; Berthele, A.; Hoshi, M.M.; Zimmer, C.; Hemmer, B.; Mühlau, M. Atrophy and structural variability of the upper cervical cord in early multiple sclerosis. Mult. Scler. 2015, 21, 875–884. [Google Scholar] [CrossRef]
- Casserly, C.; Seyman, E.E.; Alcaide-Leon, P.; Guenette, M.; Lyons, C.; Sankar, S.; Svendrovski, A.; Baral, S.; Oh, J. Spinal Cord Atrophy in Multiple Sclerosis: A Systematic Review and Meta-Analysis. J. Neuroimaging 2018, 28, 556–586. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, R.; Papinutto, N.; Zhu, A.H.; Lobach, I.V.; Bevan, C.J.; Bucci, M.; Castellano, A.; Gelfand, J.M.; Graves, J.S.; Green, A.J.; et al. Association Between Thoracic Spinal Cord Gray Matter Atrophy and Disability in Multiple Sclerosis. JAMA Neurol. 2015, 72, 897–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agosta, F.; Pagani, E.; Caputo, D.; Fillippi, M. Associations between cervical cord gray matter damage and disability in patients with multiple sclerosis. Arch. Neurol. 2007, 64, 1302–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlaeger, R.; Papinutto, N.; Panara, V.; Bevan, C.; Lobach, I.V.; Bucci, M.; Caverzasi, E.; Gelfand, J.M.; Green, A.J.; Jordan, K.M.; et al. Spinal cord gray matter atrophy correlates with multiple sclerosis disability. Ann. Neurol. 2014, 76, 568–580. [Google Scholar] [CrossRef]
- Tsagkas, C.; Magon, S.; Gaetano, L.; Pezold, S.; Naegelin, Y.; Amann, M.; Stippich, C.; Cattin, P.; Wuerfel, J.; Bieri, O.; et al. Spinal cord volume loss: A marker of disease progression in multiple sclerosis. Neurology 2018, 91, e349–e358. [Google Scholar] [CrossRef] [Green Version]
- Kearney, H.; Rocca, M.A.; Valsasina, P.; Balk, L.; Sastre-Garriga, J.; Reinhardt, J.; Ruggieri, J.; Rovira, A.; Stippich, C.; Kappos, L.; et al. Magnetic resonance imaging correlates of physical disability in relapse onset multiple sclerosis of long disease duration. Mult. Scler. 2014, 20, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Sotirchos, E.S.; Saidha, S.; Whetstone, A.; Chen, M.; Newsome, S.D.; Zackowski, K.; Balcer, L.J.; Frohman, E.; Prince, J.; et al. Relationships between quantitative spinal cord MRI and retinal layers in multiple sclerosis. Neurology 2015, 84, 720–728. [Google Scholar] [CrossRef] [Green Version]
- Valsasina, P.; Agosta, F.; Absinta, M.; Sala, S.; Caputo, D.; Filippi, M. Cervical cord functional MRI changes in relapse-onset MS patients. J. Neurol. Neurosurg. Psychiatry 2010, 81, 405–408. [Google Scholar] [CrossRef]
- Lukas, C.; Knol, D.L.; Sombekke, M.H.; Bellenberg, B.; Hahn, H.K.; Popescu, V.; Weier, K.; Radue, E.W.; Gass, A.; Kappos, L.; et al. Cervical spinal cord volume loss is related to clinical disability progression in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2015, 86, 410–418. [Google Scholar] [CrossRef]
- Rocca, M.A.; Valsasina, P.; Damjanovic, D.; Horsfield, M.A.; Mesaros, S.; Stosic-Opincal, T.; Drulovic, J.; Filippi, M. Voxel-wise mapping of cervical cord damage in multiple sclerosis patients with different clinical phenotypes. J. Neurol. Neurosurg. Psychiatry 2013, 84, 35–41. [Google Scholar] [CrossRef]
- Lin, X.; Blumhardt, L.D.; Constantinescu, C.S. The relationship of brain and cervical cord volume to disability in clinical subtypes of multiple sclerosis: A three-dimensional MRI study. Acta Neurol. Scand. 2003, 108, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, C.P.; Deluca, G.C.; Bö, L.; Owens, T.; Lowe, J.; Esiri, M.M.; Evangelou, N. Spinal cord neuronal pathology in multiple sclerosis. Brain Pathol. 2009, 19, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Sastre-Garriga, J.; Filippi, M.; Khaleeli, Z.; Téllez, N.; Vellinga, M.M.; Tur, C.; Brochet, B.; Barkhof, F.; Rovaris, M.; et al. Primary progressive multiple sclerosis diagnostic criteria: A reappraisal. Mult. Scler. 2009, 15, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Furby, J.; Hayton, T.; Smith, K.J.; Altmann, D.R.; Brenner, R.; Chataway, J.; Hughes, R.A.; Miller, D.H. Lamotrigine for neuroprotection in secondary progressive multiple sclerosis: A randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Neurol. 2010, 9, 681–688. [Google Scholar] [CrossRef]
- Cawley, N.; Tur, C.; Prados, F.; Plantone, D.; Kearney, H.; Abdel-Aziz, K.; Ourselin, S.; Wheeler-Kingshott, C.A.G.; Miller, D.H.; Thompson, A.J.; et al. Spinal cord atrophy as a primary outcome measure in phase II trials of progressive multiple sclerosis. Mult. Scler. 2018, 24, 932–941. [Google Scholar] [CrossRef]
- Evangelou, N.; DeLuca, G.C.; Owens, T.; Esiri, M.M. Pathological study of spinal cord atrophy in multiple sclerosis suggests limited role of local lesions. Brain 2005, 128, 29–34. [Google Scholar] [CrossRef]
- Kearney, H.; Miszkiel, K.A.; Yiannakas, M.C.; Ciccarelli, O.; Miller, D.H. A pilot MRI study of white and grey matter involvement by multiple sclerosis spinal cord lesions. Mult. Scler. Relat. Disord. 2013, 2, 103–108. [Google Scholar] [CrossRef]
- Philpott, C.; Brotchie, P. Comparison of MRI sequences for evaluation of multiple sclerosis of the cervical spinal cord at 3 T. Eur. J. Radiol. 2011, 80, 780–785. [Google Scholar] [CrossRef]
- Calabrese, M.; De Stefano, N.; Atzori, M.; Bernardi, V.; Mattisi, I.; Barachino, L.; Rinaldi, L.; Morra, A.; McAuliffe, M.M.; Perini, P.; et al. Detection of cortical inflammatory lesions by double inversion recovery magnetic resonance imaging in patients with multiple sclerosis. Arch. Neurol. 2007, 64, 1416–1422. [Google Scholar] [CrossRef] [Green Version]
- Bot, J.C.; Barkhof, F.; Lycklama à Nijeholt, G.J.; Bergers, E.; Polman, C.H.; Adèr, H.J.; Castelijns, J.A. Comparison of a conventional cardiac-triggered dual spin-echo and a fast STIR sequence in detection of spinal cord lesions in multiple sclerosis. Eur. Radiol. 2000, 10, 753–758. [Google Scholar] [CrossRef]
- Sethi, V.; Yousry, T.A.; Muhlert, N.; Ron, M.; Golay, X.; Wheeler-Kingshott, C.; Miller, D.H.; Chard, D.T. Improved detection of cortical MS lesions with phase-sensitive inversion recovery MRI. J. Neurol. Neurosurg. Psychiatry 2012, 83, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Nair, G.; Absinta, M.; Reich, D.S. Optimized T1-MPRAGE sequence for better visualization of spinal cord multiple sclerosis lesions at 3T. AJNR Am. J. Neuroradiol. 2013, 34, 2215–2222. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, C.P.; Bö, L.; Owens, T.; Lowe, J.; Esiri, M.M.; Evangelou, N. Spinal cord gray matter demyelination in multiple sclerosis-a novel pattern of residual plaque morphology. Brain Pathol. 2006, 16, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D.; Baier, M.; Cutter, G. Effect of relapses on development of residual deficit in multiple sclerosis. Neurology 2003, 61, 1528–1532. [Google Scholar] [CrossRef] [PubMed]
- Koch-Henriksen, N.; Thygesen, L.C.; Sørensen, P.S.; Migyari, M. Worsening of disability caused by relapses in multiple sclerosis: A different approach. Mult. Scler. Relat. Disord. 2019, 32, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkovich, R.R. Acute Multiple Sclerosis Relapse. Continuum (Minneap Minn) 2016, 22, 799–814. [Google Scholar] [CrossRef]
- Goodin, D.S. Glucocorticoid treatment of multiple sclerosis. Handb. Clin. Neurol. 2014, 122, 455–464. [Google Scholar]
- Stoppe, M.; Busch, M.; Krizek, L.; Then Bergh, F. Outcome of MS relapses in the era of disease-modifying therapy. BMC Neurol. 2017, 17, 151. [Google Scholar] [CrossRef] [Green Version]
- Schröder, A.; Linker, R.A.; Gold, R. Plasmapheresis for neurological disorders. Expert Rev. Neurother. 2009, 9, 1331–1339. [Google Scholar] [CrossRef]
- Pfeuffer, S.; Rolfes, L.; Bormann, E.; Sauerland, C.; Ruck, T.; Schilling, M.; Melzer, N.; Brand, M.; Pul, R.; Kleinschnitz, C.; et al. Comparing Plasma Exchange to Escalated Methyl Prednisolone in Refractory Multiple Sclerosis Relapses. J. Clin. Med. 2019, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Keegan, M.; Pineda, A.A.; McClelland, R.L.; Darby, C.H.; Rodriguez, M.; Weinshenker, B.G. Plasma exchange for severe attacks of CNS demyelination: Predictors of response. Neurology 2002, 58, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Derfuss, T.; Mehling, M.; Papadopoulou, A.; Bar-Or, A.; Cohen, J.A.; Kappos, L. Advances in oral immunomodulating therapies in relapsing multiple sclerosis. Lancet Neurol. 2020, 19, 336–347. [Google Scholar] [CrossRef]
- Ontaneda, D.; Tallantyre, E.; Kalincik, T.; Planchon, S.M.; Evangelou, N. Early highly effective versus escalation treatment approaches in relapsing multiple sclerosis. Lancet Neurol. 2019, 18, 973–980. [Google Scholar] [CrossRef]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Gaitán, M.I.; Ysrraelit, M.C.; Fiol, M.P. Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain 2017, 140, 527–546. [Google Scholar] [CrossRef] [Green Version]
- Young, N.P.; Weinshenker, B.G.; Lucchinetti, C.F. Acute disseminated encephalomyelitis: Current understanding and controversies. Semin. Neurol. 2008, 28, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Tenembaum, S.; Chitnis, T.; Ness, J.; Hahn, J.S.; International Pediatric MS Study Group. Acute disseminated encephalomyelitis. Neurology 2007, 68, S23–S36. [Google Scholar] [CrossRef]
- Tenembaum, S.N. Acute disseminated encephalomyelitis. Handb. Clin. Neurol. 2013, 112, 1253–1262. [Google Scholar]
- de Mol, C.L.; Wong, Y.Y.M.; van Pelt, E.D.; Ketelslegers, I.A.; Bakker, D.P.; Boon, M.; Braun, K.P.J.; van Dijk, K.G.J.; Eikelenboom, M.J.; Engelen, M.; et al. Incidence and outcome of acquired demyelinating syndromes in Dutch children: Update of a nationwide and prospective study. J. Neurol. 2018, 265, 1310–1319. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Torisu, H.; Kira, R.; Ishizaki, Y.; Sakai, Y.; Sanefuji, M.; Ichiyama, T.; Oka, A.; Kishi, T.; Kimura, S.; et al. A nationwide survey of pediatric acquired demyelinating syndromes in Japan. Neurology 2016, 87, 2006–2015. [Google Scholar] [CrossRef] [Green Version]
- Xiong, C.H.; Yan, Y.; Liao, Z.; Peng, S.H.; Wen, H.R.; Zhang, Y.X.; Chen, S.H.; Li, J.; Chen, H.Y.; Feng, X.W.; et al. Epidemiological characteristics of acute disseminated encephalomyelitis in Nanchang, China: A retrospective study. BMC Public Health 2014, 14, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karussis, D.; Petrou, P. The spectrum of post-vaccination inflammatory CNS demyelinating syndromes. Autoimmun. Rev. 2014, 13, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Tenembaum, S.; Chamoles, N.; Fejerman, N. Acute disseminated encephalomyelitis: A long-term follow-up study of 84 pediatric patients. Neurology 2002, 59, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, R.S.; Oldstone, M.B. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: Mechanism for autoimmunity. Science 1985, 230, 1043–1045. [Google Scholar] [CrossRef]
- Smyk, D.S.; Alexander, A.K.; Walker, M.; Walker, M. Acute disseminated encephalomyelitis progressing to multiple sclerosis: Are infectious triggers involved? Immunol. Res. 2014, 60, 16–22. [Google Scholar] [CrossRef]
- Krupp, L.B.; Tardieu, M.; Amato, M.P.; Banwell, B.; Chitnis, T.; Dale, R.C.; Ghezzi, A.; Hintzen, R.; Kornberg, A.; Poh, D.; et al. International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: Revisions to the 2007 definitions. Mult. Scler. J. 2013, 19, 1261–1267. [Google Scholar] [CrossRef]
- Cole, J.; Evans, E.; Mwangi, M.; Mar, S. Acute Disseminated Encephalomyelitis in Children: An Updated Review Based on Current Diagnostic Criteria. Pediatr. Neurol. 2019, 100, 26–34. [Google Scholar] [CrossRef]
- Flanagan, E.P. Autoimmune myelopathies. Handb. Clin. Neurol. 2016, 133, 327–351. [Google Scholar]
- Callen, D.J.A.; Shroff, M.M.; Branson, H.M.; Li, D.K.; Lotze, T.; Stephens, D.; Banwell, B.L. Role of MRI in the differentiation of ADEM from MS in children. Neurology 2009, 72, 968–973. [Google Scholar] [CrossRef]
- Baumann, M.; Sahin, K.; Lechner, C.; Wendel, E.M.; Lechner, C.; Behring, B.; Blaschek, A.; Diepold, K.; Eisenkölbl, A.; Fluss, J.; et al. Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein. J. Neurol. Neurosurg. Psychiatry 2015, 86, 265–272. [Google Scholar] [CrossRef]
- Ketelslegers, I.A.; Visser, I.; Neuteboom, R.F.; Boon, M.; Catsman-Berrevoets, C.E.; Hintzen, R.Q. Disease course and outcome of acute disseminated encephalomyelitis is more severe in adults than in children. Mult. Scler. J. 2011, 17, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Leake, J.A.D.; Albani, S.; Kao, A.S.; Senac, M.O.; Billman, G.F.; Nespeca, M.P.; Paulino, A.D.; Quintela, E.R.; Sawyer, M.H.; Bradley, J.S. Acute disseminated encephalomyelitis in childhood: Epidemiologic, clinical and laboratory features. Pediatr. Infect. Dis. J. 2004, 23, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Hung, P.C.; Wang, H.S.; Chou, M.L.; Lin, K.L.; Hsieh, M.Y.; Wong, A.M.C. Acute disseminated encephalomyelitis in children: A single institution experience of 28 patients. Neuropediatrics 2012, 43, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Pavone, P.; Pettoello-Mantovano, M.; Le Pira, A.; Giardino, I.; Pulvirenti, A.; Giugno, R.; Parano, E.; Polizzi, A.; Distefano, A.; Ferro, A.; et al. Acute disseminated encephalomyelitis: A long-term prospective study and meta-analysis. Neuropediatrics 2010, 41, 246–255. [Google Scholar] [CrossRef]
- Erol, I.; Ozkale, Y.; Alkan, O.; Alehan, F. Acute disseminated encephalomyelitis in children and adolescents: A single center experience. Pediatr. Neurol. 2013, 49, 266–273. [Google Scholar] [CrossRef]
- Mikaeloff, Y.; Caridade, G.; Husson, B.; Suissa, S.; Tardieu, M.; Neuropediatric KIDSEP Study Group of the French Neuropediatric Society. Acute disseminated encephalomyelitis cohort study: Prognostic factors for relapse. Eur. J. Paediatr. Neurol. 2007, 11, 90–95. [Google Scholar] [CrossRef]
- Pohl, D.; Alper, G.; Van Haren, K.; Kornberg, A.J.; Lucchinetti, C.F.; Tenembaum, S.; Belman, A.L. Acute disseminated encephalomyelitis: Updates on an inflammatory CNS syndrome. Neurology 2016, 87, S38–S45. [Google Scholar] [CrossRef]
- Verhey, L.H.; Branson, H.M.; Shroff, M.M.; Callen, D.J.; Sled, J.G.; Narayanan, S.; Sadovnick, A.D.; Bar-Or, A.; Arnold, D.L.; Marrie, R.A.; et al. MRI parameters for prediction of multiple sclerosis diagnosis in children with acute CNS demyelination: A prospective national cohort study. Lancet Neurol. 2011, 10, 1065–1073. [Google Scholar] [CrossRef]
- Lin, C.H.; Jeng, J.S.; Hsieh, S.T.; Yip, P.K.; Wu, R.M. Acute disseminated encephalomyelitis: A follow-up study in Taiwan. J. Neurol. Neurosurg. Psychiatry 2007, 78, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.; Gorman, M.; Rensel, M.; Austin, T.E.; Hertz, D.P.; Kuntz, N.L. Network of Pediatric Multiple Sclerosis Centers of Excellence of the National Multiple Sclerosis Society. Management of Pediatric Central Nervous System Demyelinating Disorders: Consensus of United States Neurologists. J. Child. Neurol. 2011, 26, 675–682. [Google Scholar] [CrossRef]
- Dale, R.C. Acute disseminated encephalomyelitis, multiphasic disseminated encephalomyelitis and multiple sclerosis in children. Brain 2000, 123, 2407–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, D.S.; Melvin, J.J.; Kothare, S.V.; Valencia, I.; Hardison, H.H.; Yum, S.; Faerber, E.N.; Legido, A. Acute disseminated encephalomyelitis in children: Discordant neurologic and neuroimaging abnormalities and response to plasmapheresis. Pediatrics 2005, 116, 431–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, D.; Tenembaum, S. Treatment of acute disseminated encephalomyelitis. Curr. Treat. Options Neurol. 2012, 14, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Wildemann, B. The history of neuromyelitis optica. J. Neuroinflamm. 2013, 10, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, P.V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Wingerchuk, D.M.; Lennon, V.A.; Pittock, S.J.; Lucchinetti, C.F.; Weinshenker, B.G. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006, 66, 1485–1489. [Google Scholar] [CrossRef] [Green Version]
- Wingerchuk, D.M.; Lennon, V.A.; Lucchinetti, C.F.; Pittock, S.J.; Weinshenker, B.G. The spectrum of neuromyelitis optica. Lancet Neurol. 2007, 6, 805–815. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef]
- Saadoun, S.; Waters, P.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 2010, 133, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Ratelade, J.; Asavapanumas, N.; Ritchie, A.M.; Wemlinger, S.; Bennett, J.L.; Verkman, A.S. Involvement of antibody-dependent cell-mediated cytotoxicity in inflammatory demyelination in a mouse model of neuromyelitis optica. Acta Neuropathol. 2013, 126, 699–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradl, M.; Misu, T.; Takahashi, T.; Watanabe, M.; Mader, S.; Reindl, M.; Adzemovic, M.; Bauer, J.; Berger, T.; Fujihara, K.; et al. Neuromyelitis optica: Pathogenicity of patient immunoglobulin in vivo. Ann. Neurol. 2009, 66, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Chang, V.T.W.; Chang, H.M. Review: Recent advances in the understanding of the pathophysiology of neuromyelitis optica spectrum disorder. Neuropathol. Appl. Neurobiol. 2020, 46, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.F.; Mandler, R.N.; McGavern, D.; Bruck, W.; Gleich, G.; Ransohoff, R.M.; Trebst, C.; Weinshenker, B.; Wingerchuk, D.; Parisi, J.E.; et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 2002, 125 Pt 7, 1450–1461. [Google Scholar] [CrossRef] [Green Version]
- Misu, T.; Fujihara, K.; Kakita, A.; Konno, H.; Nakamura, M.; Watanabe, S.; Takahashi, T.; Nakashima, I.; Takahashi, H.; Itoyama, Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: Distinction from multiple sclerosis. Brain 2007, 130, 1224–1234. [Google Scholar] [CrossRef] [Green Version]
- Pittock, S.J.; Lennon, V.A.; Krecke, K.; Wingerchuk, D.M.; Lucchinetti, C.F.; Weinshenker, B.G. Brain abnormalities in neuromyelitis optica. Arch. Neurol. 2006, 63, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Hinson, S.R.; Roemer, S.F.; Lucchinetti, C.F.; Fryer, J.P.; Kryzer, T.J.; Chamberlain, J.L.; Howe, C.L.; Pittock, S.J.; Lennon, V.A. Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down- Regulating EAAT2. J. Exp. Med. 2008, 205, 2473–2481. [Google Scholar] [CrossRef]
- Papadopoulos, M.; Verkman, A.S. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2009, 53, 820–833. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, J.F.; Hoffman, B.M.; Tyor, W.R. CNS inflammatory demyelinating disorders: MS, NMOSD and MOG antibody associated disease. J. Investig. Med. 2020, 68, 321–330. [Google Scholar] [CrossRef]
- Jarius, S.; Ruprecht, K.; Wildemann, B.; Kuempfel, T.; Ringelstein, M.; Geis, C.; Kleiter, I.; Kleinschnitz, C.; Berthele, A.; Brettschneider, J.; et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: A multicentre study of 175 patients. J. Neuroinflamm. 2012, 9, 14. [Google Scholar] [CrossRef]
- Ghezzi, A.; Bergamaschi, R.; Martinelli, V.; Trojano, M.; Tola, M.R.; Merelli, E.; Mancardi, L.; Gallo, P.; Filippi, M.; Zaffaroni, M.; et al. Clinical characteristics, course and prognosis of relapsing Devic’s Neuromyelitis Optica. J. Neurol. 2004, 251, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Hogancamp, W.F.; O’Brien, P.C.; Weinshenker, BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 1999, 53, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Yonezu, T.; Ito, S.; Mori, M.; Ogawa, Y.; Makino, T.; Uzawa, A.; Kuwabara, S. Bright spotty lesions on spinal magnetic resonance imaging differentiate neuromyelitis optica from multiple sclerosis. Mult. Scler. J. 2014, 20, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, E.P.; Kaufmann, T.J.; Krecke, K.N.; Aksamit, A.J.; Pittock, S.J.; Keegan, B.M.; Giannini, C.; Weinshenker, B.G. Discriminating long myelitis of neuromyelitis optica from sarcoidosis. Ann. Neurol. 2016, 79, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Iorio, R.; Damato, V.; Mirabella, M.; Evoli, A.; Marti, A.; Plantone, D.; Frisullo, G.; Batocchi, A.P. Distinctive clinical and neuroimaging characteristics of longitudinally extensive transverse myelitis associated with aquaporin-4 autoantibodies. J. Neurol. 2013, 260, 2396–2402. [Google Scholar] [CrossRef]
- Flanagan, E.P.; Weinshenker, B.G.; Krecke, K.N.; Lennon, V.A.; Lucchinetti, C.F.; McKeon, A.; Wingerchuk, D.M.; Shuster, E.A.; Jiao, Y.; Horta, E.S.; et al. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol. 2015, 72, 81–87. [Google Scholar] [CrossRef]
- Kim, S.H.; Huh, S.Y.; Kim, W.; Park, M.S.; Ahn, S.E.; Cho, J.Y.; Kim, B.J.; Kim, H.J. Clinical characteristics and outcome of multiple sclerosis in Korea: Does multiple sclerosis in Korea really differ from that in the Caucasian populations? Mult. Scler. J. 2013, 19, 1493–1498. [Google Scholar] [CrossRef]
- Scott, T.F. Nosology of idiopathic transverse myelitis syndromes. Acta Neurol. Scand. 2007, 115, 371–376. [Google Scholar] [CrossRef]
- Asgari, N.; Skejoe, H.P.B.; Lillevang, S.T.; Steenstrup, T.; Stenager, E.; Kyvik, K.O. Modifications of longitudinally extensive transverse myelitis and brainstem lesions in the course of neuromyelitis optica (NMO): A population-based, descriptive study. BMC Neurol. 2013, 13, 33. [Google Scholar] [CrossRef]
- Hamid, S.H.M.; Elsone, L.; Mutch, K.; Solomon, T.; Jacob, A. The impact of 2015 neuromyelitis optica spectrum disorders criteria on diagnostic rates. Mult. Scler. 2017, 23, 228–233. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, Y.; Schoonheim, M.M.; Zhang, N.; Fan, M.; Su, L.; Shen, Y.; Yan, Y.; Yang, L.; Wang, Q.; et al. Structural MRI substrates of cognitive impairment in neuromyelitis optica. Neurology 2015, 85, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Ventura, R.E.; Kister, I.; Chung, S.; Babb, J.S.; Shepherd, T.M. Cervical spinal cord atrophy in NMOSD without a history of myelitis or MRI-visible lesions. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, J.W.; Jeong, I.H.; Joung, A.; Kim, S.H.; Kim, H.J. Evaluation of the 2015 diagnostic criteria for neuromyelitis optica spectrum disorder. Neurology 2016, 86, 1772–1779. [Google Scholar] [CrossRef] [PubMed]
- Marignier, R.; Bernard-Valnet, R.; Giraudon, P.; Collongues, N.; Papeix, C.; Zéphir, H.; Cavillon, G.; Rogemond, V.; Casey, R.; Frangoulis, B.; et al. Aquaporin-4 antibody-negative neuromyelitis optica: Distinct assay sensitivity-dependent entity. Neurology 2013, 80, 2194–2200. [Google Scholar] [CrossRef]
- Pittock, S.J.; Lennon, V.A.; Bakshi, N.; Shen, S.; McKeon, A.; Quach, H.; Briggs, F.B.S.; Bernstein, A.L.; Schaefer, C.A.; Barcellos, L.F. Seroprevalence of aquaporin-4-IgG in a northern California population representative cohort of multiple sclerosis. JAMA Neurol. 2014, 71, 1433–1436. [Google Scholar] [CrossRef] [Green Version]
- Jarius, S.; Wildemann, B.; Paul, F. Neuromyelitis optica: Clinical features, immunopathogenesis and treatment. Clin. Exp. Immunol. 2014, 176, 149–164. [Google Scholar] [CrossRef]
- van Pelt, E.D.; Wong, Y.Y.M.; Ketelslegers, I.A.; Hamann, D.; Hintzen, R.Q. Neuromyelitis optica spectrum disorders: Comparison of clinical and magnetic resonance imaging characteristics of AQP4-IgG versus MOG-IgG seropositive cases in the Netherlands. Eur. J. Neurol. 2016, 23, 580–587. [Google Scholar] [CrossRef]
- Hamid, S.H.M.; Whittam, D.; Mutch, K.; Linaker, S.; Solomon, T.; Das, K.; Bhojak, M.; Jacob, A. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J. Neurol. 2017, 264, 2088–2094. [Google Scholar] [CrossRef] [Green Version]
- Höftberger, R.; Sepulveda, M.; Armangue, T.; Blanco, Y.; Rostásy, K.; Cobo Calvo, A.; Olascoaga, J.; Ramió-Torrentà, L.; Reindl, M.; Benito-León, J.; et al. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult. Scler. 2015, 21, 866–874. [Google Scholar] [CrossRef]
- Majed, M.; Fryer, J.P.; McKeon, A.; Lennon, V.A.; Pittock, S.J. Clinical utility of testing AQP4-IgG in CSF: Guidance for physicians. Neurol. Neuroimmunol. NeuroInflamm. 2016, 3, e231. [Google Scholar] [CrossRef] [Green Version]
- Javed, A.; Balabanov, R.; Arnason, B.G.W.; Kelly, T.J.; Sweiss, N.J.; Pytel, P.; Walsh, R.; Blair, E.A.; Stemer, A.; Lazzaro, M.; et al. Minor salivary gland inflammation in Devic’s disease and longitudinally extensive myelitis. Mult. Scler. 2008, 14, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Wandinger, K.P.; Stangel, M.; Witte, T.; Venables, P.; Charles, P.; Jarius, S.; Wildemann, B.; Probst, C.; Iking-Konert, C.; Schneider, M. Autoantibodies against aquaporin-4 in patients with neuropsychiatric systemic lupus erythematosus and primary Sjögren’s syndrome. Arthritis Rheumatol. 2010, 62, 1198–1200. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Paul, F.; Franciotta, D.; Ruprecht, K.; Ringelstein, M.; Bergamaschi, R.; Rommer, P.; Kleiter, I.; Stich, O.; Reuss, R.; et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: Results from 211 lumbar punctures. J. Neurol. Sci. 2011, 306, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Weinshenker, B.G.; Wingerchuk, D.M. Neuromyelitis Spectrum Disorders. Mayo Clin. Proc. 2017, 92, 663–679. [Google Scholar] [CrossRef] [Green Version]
- Kessler, R.A.; Mealy, M.A.; Levy, M. Treatment of Neuromyelitis Optica Spectrum Disorder: Acute, Preventive, and Symptomatic. Curr. Treat. Options Neurol. 2016, 18, 2. [Google Scholar] [CrossRef] [Green Version]
- Trebst, C.; Jarius, S.; Berthele, A.; Paul, F.; Schippling, S.; Wildemann, B.; Borisow, N.; Kleiter, I.; Aktas, O.; Kümpfel, T.; et al. Update on the diagnosis and treatment of neuromyelitis optica: Recommendations of the Neuromyelitis Optica Study Group (NEMOS). J. Neurol. 2014, 261, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Palace, J.; Leite, I.; Jacob, A. A practical guide to the treatment of neuromyelitis optica. Pract. Neurol. 2012, 12, 209–214. [Google Scholar] [CrossRef]
- Magaña, S.M.; Keegan, B.M.; Weinshenker, B.G.; Erickson, B.J.; Pittock, S.J.; Lennon, V.A.; Rodriguez, M.; Thomsen, K.; Weigand, S.; Mandrekar, J.; et al. Beneficial Plasma Exchange Response in CNS Inflammatory Demyelination. Arch. Neurol. 2012, 68, 870–878. [Google Scholar] [CrossRef] [Green Version]
- Reeves, H.M.; Winters, J.L. The mechanisms of action of plasma exchange. Br. J. Haematol. 2014, 164, 342–351. [Google Scholar] [CrossRef]
- Bonnan, M.; Valentino, R.; Debeugny, S.; Merle, H.; Fergé, J.L.; Mehdaoui, H.; Cabre, P. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J. Neurol. Neurosurg. Psychiatry 2018, 89, 346–351. [Google Scholar] [CrossRef]
- Lim, Y.M.; Pyun, S.Y.; Kang, B.H.; Kim, J.; Kim, K.K. Factors associated with the effectiveness of plasma exchange for the treatment of NMO-IgG-positive neuromyelitis optica spectrum disorders. Mult. Scler. J. 2013, 19, 1216–1218. [Google Scholar] [CrossRef] [PubMed]
- Bonnan, M.; Valentino, R.; Olindo, S.; Mehdaoui, H.; Smadja, D.; Cabre, P. Plasma exchange in severe spinal attacks associated with neuromyelitis optica spectrum disorder. Mult. Scler. 2009, 15, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Nakashima, I.; Misu, T.; Miyazawa, I.; Shiga, Y.; Fujihara, K.; Itoyama, Y. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult. Scler. 2007, 13, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N. Engl. J. Med. 2019, 381, 614–625. [Google Scholar] [CrossRef]
- Cree, B.A.C.; Bennett, J.L.; Kim, H.J.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; Fujihara, K.; Paul, F.; Cutter, G.R.; Marignier, R.; et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): A double-blind, randomised placebo-controlled phase 2/3 trial. Lancet 2019, 394, 1352–1363. [Google Scholar] [CrossRef]
- Yamamura, T.; Kleiter, I.; Fujihara, K.; Palace, J.; Greenberg, B.; Zakrzewska-Pniewska, B.; Patti, F.; Tsai, C.P.; Saiz, A.; Yamazaki, H.; et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N. Engl. J. Med. 2019, 381, 2114–2124. [Google Scholar] [CrossRef]
- Reindl, M.; Linington, C.; Brehm, U.; Egg, R.; Dilitz, E.; Deisenhammer, F.; Poewe, W.; Berger, T. Antibodies against the myelin oligodendrocyte glycoprotein and the myelin basic protein in multiple sclerosis and other neurological diseases: A comparative study. Brain 1999, 122, 2047–2056. [Google Scholar] [CrossRef] [Green Version]
- Berger, T.; Rubner, P.; Schautzer, F.; Egg, R.; Ulmer, H.; Mayringer, I.; Dilitz, E.; Deisenhammer, F.; Reindl, M. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N. Engl. J. Med. 2003, 349, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Berger, T.; Reindl, M. Lack of association between antimyelin antibodies and progression to multiple sclerosis. N. Engl. J. Med. 2007, 356, 1888–1889. [Google Scholar]
- Lampasona, V.; Franciotta, D.; Furlan, R.; Zanaboni, S.; Fazio, R.; Bonifacio, E.; Comi, G.; Martino, G. Similar low frequency of anti-MOG IgG and IgM in MS patients and healthy subjects. Neurology 2004, 62, 2092–2094. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.T.; Berger, T.; Reindl, M.; Dalton, C.M.; Fernando, K.; Keir, G.; Thompson, E.J.; Miller, D.H.; Giovannoni, G. Anti-myelin antibodies do not allow earlier diagnosis of multiple sclerosis. Mult. Scler. 2005, 11, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Hennes, E.M.; Baumann, M.; Schanda, K.; Anlar, B.; Bajer-Kornek, B.; Blaschek, A.; Brantner-Inthaler, S.; Diepold, K.; Eisenkölbl, A.; Gotwald, T.; et al. Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology 2017, 89, 900–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brilot, F.; Dale, R.C.; Selter, R.C.; Grummel, V.; Kalluri, S.R.; Aslam, M.; Busch, V.; Zhou, D.; Cepok, S.; Hemmer, B. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann. Neurol. 2009, 66, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Huppke, P.; Rostasy, K.; Karenfort, M.; Huppke, B.; Seidl, R.; Leiz, S.; Reindl, M.; Gärtner, J. Acute disseminated encephalomyelitis followed by recurrent or monophasic optic neuritis in pediatric patients. Mult. Scler. J. 2013, 19, 941–946. [Google Scholar] [CrossRef]
- O’Connor, K.C.; McLaughlin, K.A.; De Jager, P.L.; Chitnis, T.; Bettelli, E.; Xu, C.; Robinson, W.H.; Cherry, S.V.; Bar-Or, A.; Banwell, B.; et al. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat. Med. 2007, 13, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Jarius, S.; Ruprecht, K.; Kleiter, I.; Borisow, N.; Asgari, N.; Pitarokoili, K.; Pache, F.; Stich, O.; Beume, L.A.; Hümmert, M.W.; et al. MOG-IgG in NMO and related disorders: A multicenter study of 50 patients. Part 1, Frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J. Neuroinflamm. 2016, 13, 279. [Google Scholar] [CrossRef] [Green Version]
- Rostasy, K.; Mader, S.; Schanda, K.; Huppke, P.; Gärtner, J.; Kraus, V.; Karenfort, M.; Tibussek, D.; Blaschek, A.; Bajer-Kornek, B.; et al. Anti-myelin oligodendrocyte glycoprotein antibodies in pediatric patients with optic neuritis. Arch. Neurol. 2012, 69, 752–756. [Google Scholar] [CrossRef] [Green Version]
- Mader, S.; Gredler, V.; Schanda, K.; Rostasy, K.; Dujmovic, I.; Pfaller, K.; Lutterotti, A.; Jarius, S.; Di Pauli, F.; Kuenz, B.; et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J. Neuroinflamm. 2011, 8, 184. [Google Scholar] [CrossRef] [Green Version]
- Hyun, J.W.; Woodhall, M.R.; Kim, S.H.; Jeong, I.H.; Kong, B.; Kim, G.; Kim, Y.; Park, M.S.; Irani, S.R.; Waters, P.; et al. Longitudinal analysis of myelin oligodendrocyte glycoprotein antibodies in CNS inflammatory diseases. J. Neurol. Neurosurg. Psychiatry 2017, 88, 811–817. [Google Scholar] [CrossRef]
- Ramanathan, S.; Mohammad, S.; Tantsis, E.; Nguyen, T.K.; Merheb, V.; Fung, V.S.C.; White, O.B.; Broadley, S.; Lechner-Scott, J.; Vucic, S.; et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J. Neurol. Neurosurg. Psychiatry 2018, 89, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Jarius, S.; Ruprecht, K.; Kleiter, I.; Borisow, N.; Asgari, N.; Pitarokoili, K.; Pache, F.; Stich, O.; Beume, L.A.; Hümmert, M.W.; et al. MOG-IgG in NMO and related disorders: A multicenter study of 50 patients. Part 2, Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J. Neuroinflamm. 2016, 13, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepúlveda, M.; Armangué, T.; Sola-Valls, N.; Arrambide, G.; Meca-Lallana, J.E.; Oreja-Guevara, C.; Mendibe, M.; Alvarez de Arcaya, A.; Aladro, Y.; Casanova, B.; et al. Neuromyelitis optica spectrum disorders: Comparison according to the phenotype and serostatus. Neurol. Neuroimmunol. NeuroInflamm. 2016, 3, e225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurynczyk, M.; Geraldes, R.; Probert, F.; Woodhall, M.R.; Waters, P.; Tackley, G.; DeLuca, G.; Chandratre, S.; Leite, M.I.; Vincent, A.; et al. Distinct brain imaging characteristics of autoantibody-mediated CNS conditions and multiple sclerosis. Brain 2017, 140, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Paul, F.; Lana-Peixoto, M.A.; Tenembaum, S.; Asgari, N.; Palace, J.; Klawiter, E.C.; Sato, D.K.; de Seze, J.; Wuerfel, J.; et al. MRI characteristics of neuromyelitis optica spectrum disorder: An international update. Neurology 2015, 84, 1165–1173. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Woodhall, M.R.; Kim, J.S.; Kim, S.J.; Park, K.S.; Vincent, A.; Lee, K.W.; Waters, P. Antibodies to MOG in adults with inflammatory demyelinating disease of the CNS. Neurol. Neuroimmunol. NeuroInflamm. 2015, 2, e163. [Google Scholar] [CrossRef] [Green Version]
- Jarius, S.; Kleiter, I.; Ruprecht, K.; Asgari, N.; Pitarokoili, K.; Borisow, N.; Hümmert, M.W.; Trebst, C.; Pache, F.; Winkelmann, A.; et al. MOG-IgG in NMO and related disorders: A multicenter study of 50 patients. Part 3, Brainstem involvement—Frequency, presentation and outcome. J. Neuroinflamm. 2016, 13, 281. [Google Scholar] [CrossRef] [Green Version]
- Kitley, J.; Waters, P.; Woodhall, M.; Leite, M.I.; Murchison, A.; George, J.; Küker, W.; Chandratre, S.; Vincent, A.; Palace, J. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies a comparative study. JAMA Neurol. 2014, 71, 276–283. [Google Scholar] [CrossRef]
- Sato, D.K.; Callegaro, D.; Lana-Peixoto, M.A.; Waters, P.J.; de Haidar Jorge, F.M.; Takahashi, T.; Nakashima, I.; Apostolos-Pereira, S.L.; Talim, N.; Simm, R.F.; et al. Distinction between MOG antibody positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 2014, 82, 474–481. [Google Scholar] [CrossRef]
- Matthews, L.; Marasco, R.; Jenkinson, M.; Küker, W.; Luppe, S.; Leite, M.I.; Giorgio, A.; De Stefano, N.; Robertson, N.; Johansen-Berg, H.; et al. Distinction of seropositive NMO spectrum disorder and MS brain lesion distribution. Neurology 2013, 80, 1330–1337. [Google Scholar] [CrossRef] [Green Version]
- Bensi, C.; Marrodan, M.; González, A.; Chertcoff, A.; Osa Sanz, E.; Chaves, H.; Schteinschnaider, A.; Correale, J.; Farez, M.F. Brain and spinal cord lesion criteria distinguishes AQP4-positive neuromyelitis optica and MOG-positive disease from multiple sclerosis. Mult. Scler. Relat. Disord. 2018, 25, 246–250. [Google Scholar] [CrossRef]
- Cobo-Calvo, A.; Ruiz, A.; Maillart, E.; Audoin, B.; Zephir, H.; Bourre, B.; Ciron, J.; Collongues, N.; Brassat, D.; Cotton, F.; et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: The MOGADOR study. Neurology 2018, 90, e1858–e1869. [Google Scholar] [CrossRef] [PubMed]
- Chalmoukou, K.; Alexopoulos, H.; Akrivou, S.; Stathopoulos, P.; Reindl, M.; Dalakas, M.C. Anti-MOG antibodies are frequently associated with steroid-sensitive recurrent optic neuritis. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitley, J.; Woodhall, M.; Waters, P.; Leite, M.I.; Devenney, E.; Craig, J.; Palace, J.; Vincent, A. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 2012, 79, 1273–1277. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, W.; Li, X.F.; Jung, I.J.; Kim, H.J. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch. Neurol. 2011, 68, 1412–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montcuquet, A.; Collongues, N.; Papeix, C.; Zephir, H.; Audoin, B.; Laplaud, D.; Bourre, B.; Brochet, B.; Camdessanche, J.P.; Labauge, P.; et al. Effectiveness of mycophenolate mofetil as first-line therapy in AQP4-IgG, MOG-IgG, and seronegative neuromyelitis optica spectrum disorders. Mult. Scler. 2017, 23, 1377–1384. [Google Scholar] [CrossRef]
- Flanagan, E.P.; Hinson, S.R.; Lennon, V.A.; Fang, B.; Aksamit, A.J.; Morris, A.P.; Basal, E.; Honorat, J.A.; Alfugham, N.N.; Linnoila, J.J.; et al. GFAP-IgG as Biomarker of Autoimmune Astrocytopathy: Analysis of 102 Patients. Ann. Neurol. 2017, 81, 298–309. [Google Scholar] [CrossRef]
- Fang, B.; McKeon, A.; Hinson, S.R.; Kryzer, T.J.; Pittock, S.J.; Aksamit, A.J.; Lennon, V.A. Autoimmune glial fibrillary acidic protein astrocytopathy: A novel meningoencephalomyelitis. JAMA Neurol. 2016, 73, 1297–1307. [Google Scholar] [CrossRef]
- Sasaki, K.; Bean, A.; Shah, S.; Schutten, E.; Huseby, P.G.; Peters, B.; Shen, Z.T.; Vanguri, V.; Liggitt, D.; Huseby, E.S. Relapsing–Remitting Central Nervous System Autoimmunity Mediated by GFAP-Specific CD8 T Cells. J. Immunol. 2014, 192, 3029–3042. [Google Scholar] [CrossRef] [Green Version]
- Schweingruber, N.; Fischer, H.J.; Fischer, L.; van den Brandt, J.; Karabinskaya, A.; Labi, V.; Villunger, A.; Kretzschmar, B.; Huppke, P.; Simons, M.; et al. Chemokine-mediated redirection of T cells constitutes a critical mechanism of glucocorticoid therapy in autoimmune CNS responses. Acta Neuropathol. 2014, 127, 713–729. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist 2014, 20, 160–172. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunchok, A.; Zekeridou, A.; McKeon, A. Autoimmune glial fibrillary acidic protein astrocytopathy. Curr. Opin. Neurol. 2019, 32, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Iorio, R.; Damato, V.; Evoli, A.M.; Guessi, M.; Gaudino, S.; Di Lazzaro, V.; Spagni, G.; Sluijs, J.A.; Hol, E.M. Clinical and immunological characteristics of the spectrum of GFAP autoimmunity: A case series of 22 patients. J. Neurol. Neurosurg. Psychiatry 2018, 89, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Sechi, E.; Morris, P.P.; Mckeon, A.; Pittock, S.J.; Hinson, S.R.; Winshenker, B.G.; Aksamit, A.J.; Krecke, K.N.; Kaufmann, T.J.; Jolliffe, E.A.; et al. Glial fibrillary acidic protein IgG related myelitis: Characterisation and comparison with aquaporin-4-IgG myelitis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 488–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, F.; Long, Y.; Qiu, W. Autoimmune Glial Fibrillary Acidic Protein Astrocytopathy: A Review of the Literature. Front. Immunol. 2018, 9, 2802. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MS | ADEM | NMOSD | MOG-IgG Disease | GFAP-IgG Disease | |
|---|---|---|---|---|---|
| Estimated F:M ratio | 3:1 | 1:1 | 9:1 | 1.3:1 | 1:1 |
| Age * (yrs) | 30 | 6 | 37 | 33 | 40 |
| Myelitis clinical features | Sensory loss, gait impairment, weakness, sphincter involvement | Transverse myelitis | Transverse myelitis | Paraparesis, sensory symptoms and sphincter involvement | Sensory symptoms, sphincter disfunction |
| Clinical course | Relapsing (85%) or progressive (15%) | Typically monophasic (69–90%) | Relapsing (90%) | Monophasic (58%) or relapsing (42%) | Relapsing (50%), monophasic (27%) or progressive (23%) |
| Serology findings | Not relevant | Not relevant | Serum AQP4-IgG. coexistence with other systemic disease antibodies (ANA, SSA or SSB). | Serum MOG-IgG | Anti-GFAP ab + in serum or CSF (Serum Anti-AQP4-IgG and/or anti-NMDAr ab coexistence, |
| Presence of OCB | 80–95% | 0% to 29% (usually transient) | Up to 30% (usually transient) | Up to 12% | Up to 50% |
| CSF | Generally normal or mild inflammatory changes | Mild pleocytosis and increased proteins up to 62% | Pleocytosis (neutrophils and eosinophils can be found) and mild elevated proteins | Normal or slightly inflammatory changes | Marked elevation of white blood cells and elevated protein levels |
| Brain MRI | Dawson fingers, lesions perpendicular to ventricles Cortical/yuxtacortical lesions Perivenular Nodular or ring/open-ring enhancing lesions Unilateral short optic nerve enhancement | Subcortical or deep gray matter bilateral, sometimes poorly-defined Simultaneous enhancement with gadolinium | Periependimal lesions Tumefactive lesions Involvement of corticospinal tract Marked enhancement, ‘cloud like’ Bilateral, long optic nerve enhancement | Non—specific supratentorial subcortical or small deep white matter foci. Occasionally T2 lesions in brainstem, and infratentorial regions Anterior bilateral ON with perineural optic nerved enhancement | Linear radial periventricular contrast enhancement pattern |
| Spinal cord MRI | Small, peripheral, posterolateral lesions Less than 3 segments Gadolinium enhancement during acute phase | LETM or multiple short segment myelitis Edematous lesions and gadolinium enhancement in acute phase | Central LETM Edematous Necrosis or cavitation Gadolinium enhancement in acute phase | LETM or short myelitis, frequent conus medullaris involvement Linear gadolinium enhancement of the ependymal canal | LETM Central lesions |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marrodan, M.; Gaitán, M.I.; Correale, J. Spinal Cord Involvement in MS and Other Demyelinating Diseases. Biomedicines 2020, 8, 130. https://doi.org/10.3390/biomedicines8050130
Marrodan M, Gaitán MI, Correale J. Spinal Cord Involvement in MS and Other Demyelinating Diseases. Biomedicines. 2020; 8(5):130. https://doi.org/10.3390/biomedicines8050130
Chicago/Turabian StyleMarrodan, Mariano, María I. Gaitán, and Jorge Correale. 2020. "Spinal Cord Involvement in MS and Other Demyelinating Diseases" Biomedicines 8, no. 5: 130. https://doi.org/10.3390/biomedicines8050130