Structure–Activity Analysis and Molecular Docking Studies of Coumarins from Toddalia asiatica as Multifunctional Agents for Alzheimer’s Disease

 ,
,
Abstract
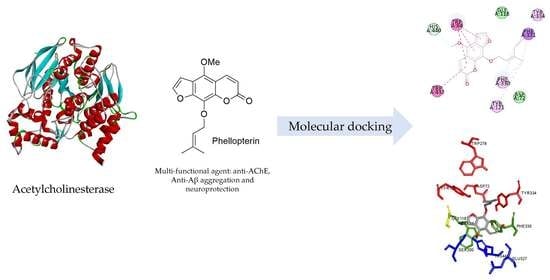
:
1. Introduction
2. Materials and Methods
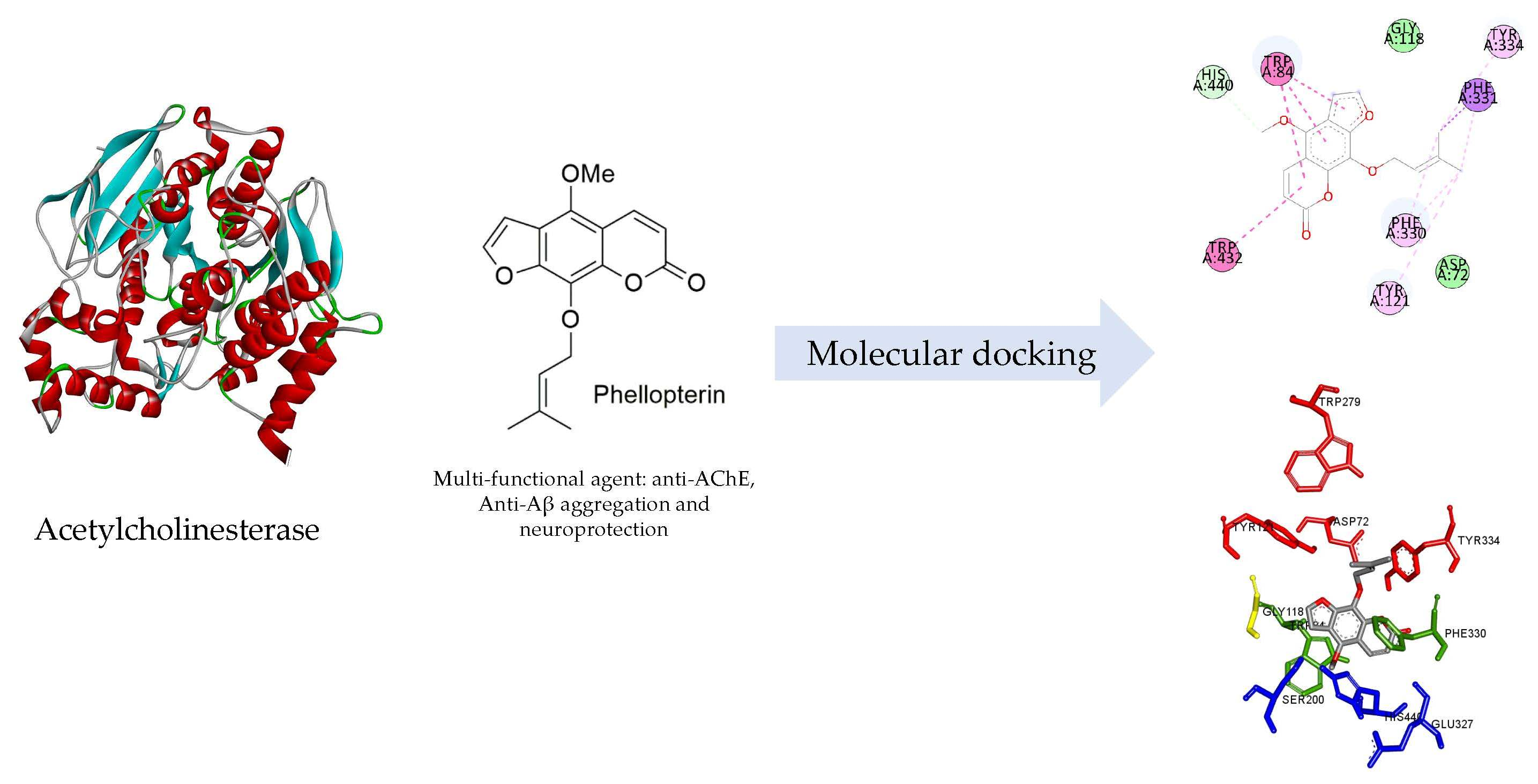
2.1. Coumarin Derivatives
2.2. Acetylcholinesterase Inhibitory Activity
2.3. Inhibition of AChE-Induced Aβ1–42 Aggregation
2.4. Inhibition of Self-Induced Aβ1–42 Aggregation
2.5. Computational Studies
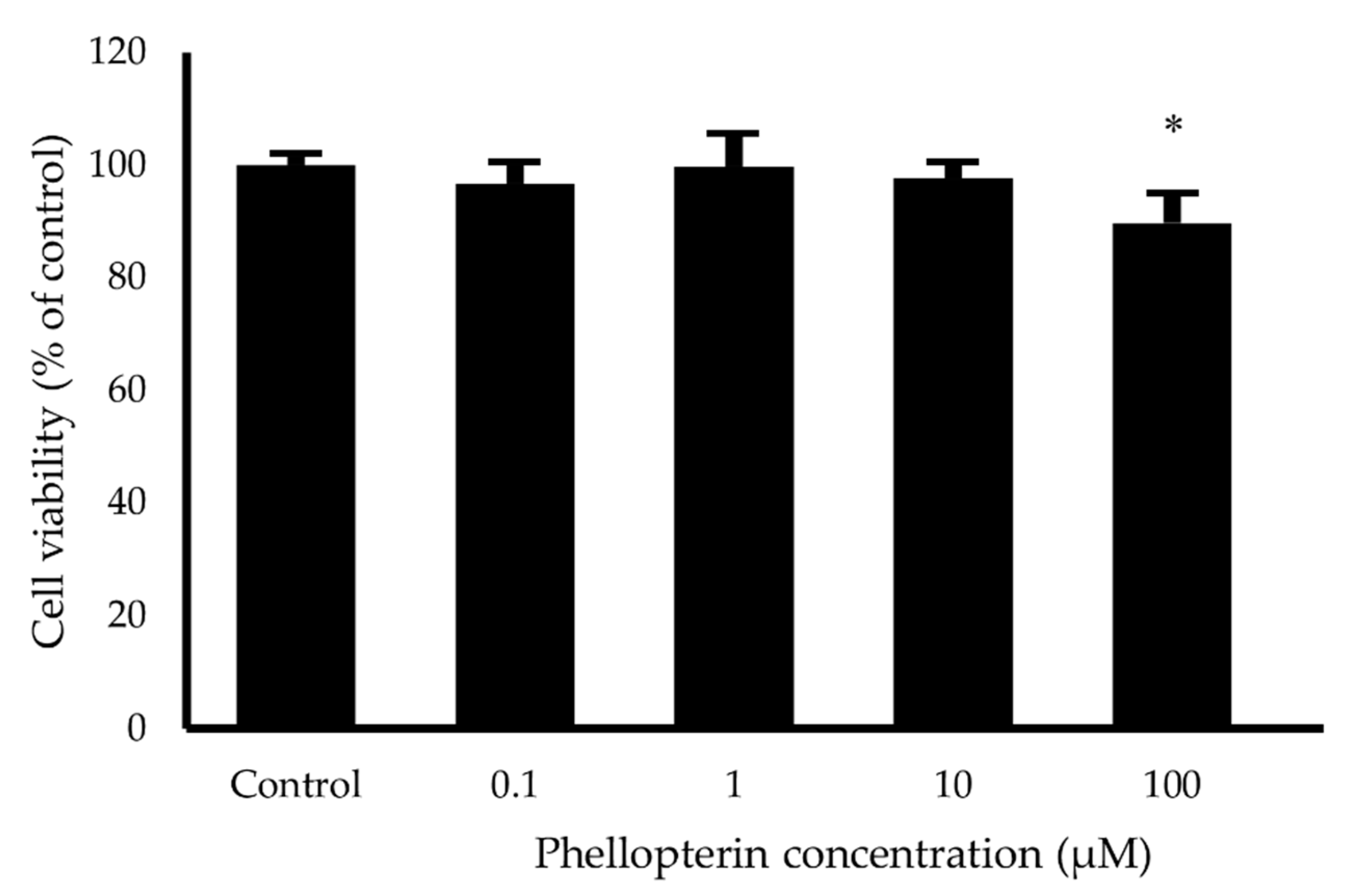
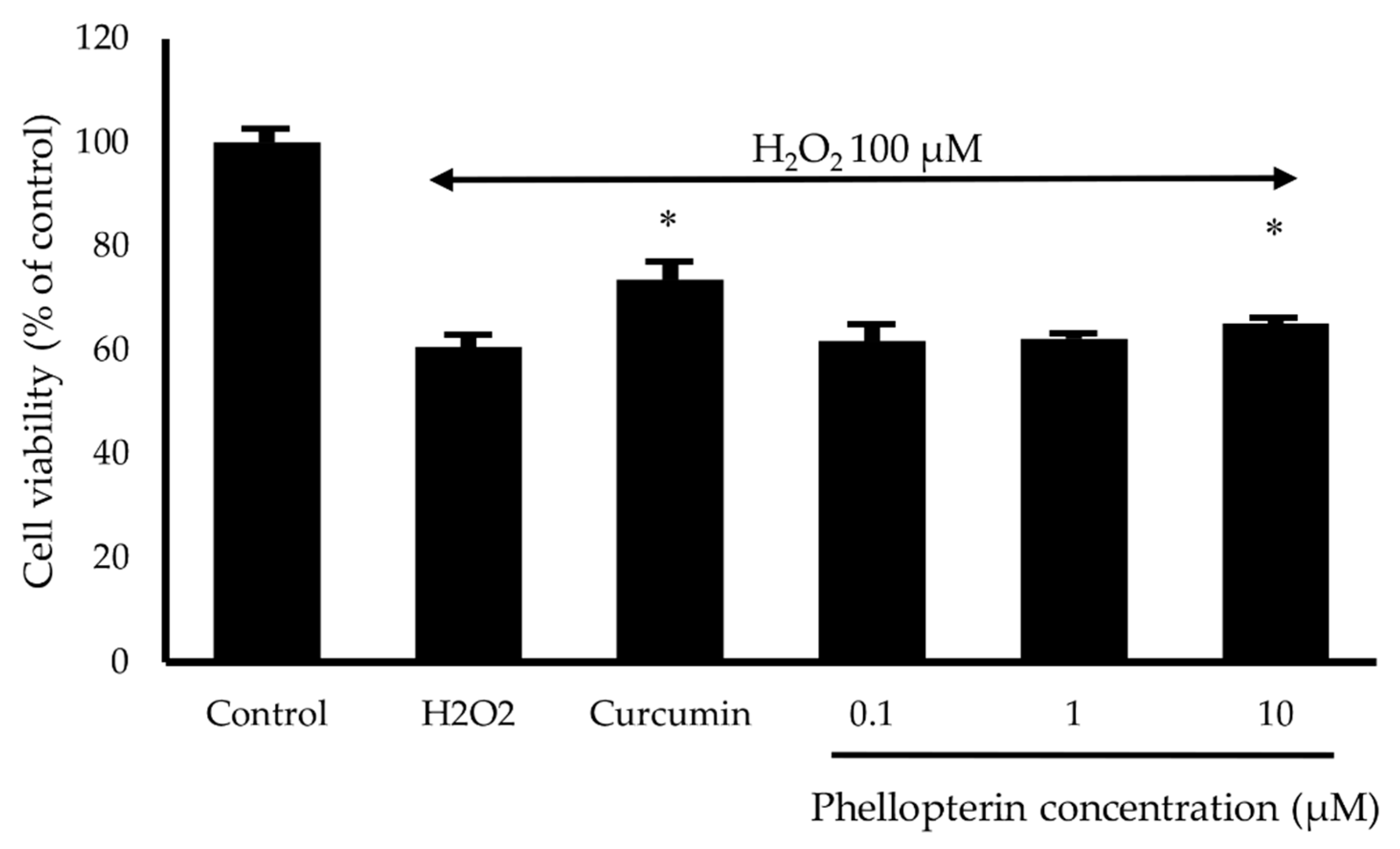
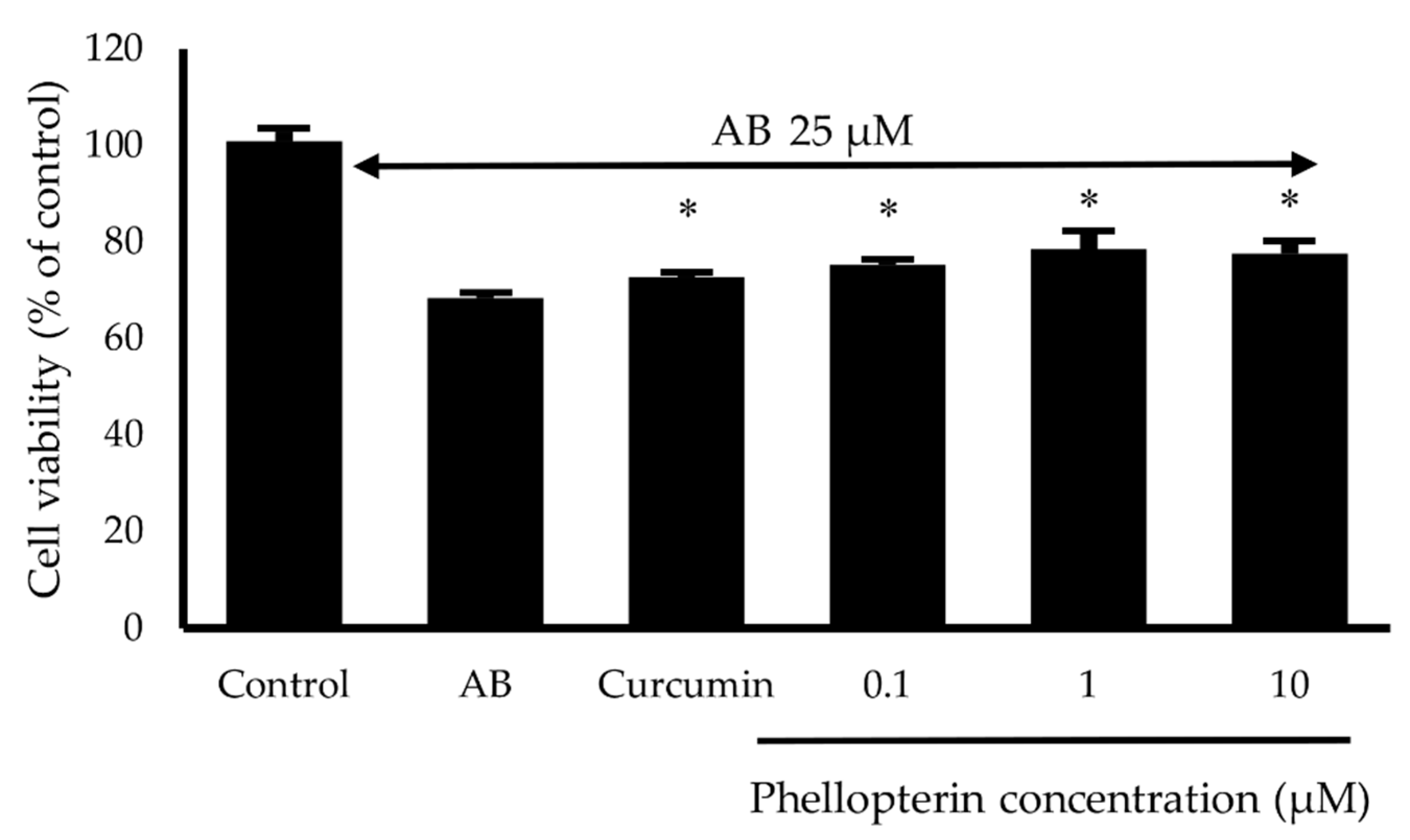
2.6. Neuroprotective Activity against Hydrogen Peroxide (H2O2) and Aβ1–42 Toxicity
3. Results and Discussion
3.1. Acetylcholinesterase Inhibition
3.2. AChE-Induced Aβ Aggregation Inhibitory Activity
3.3. Self-Induced Aβ Aggregation Inhibitory Activity
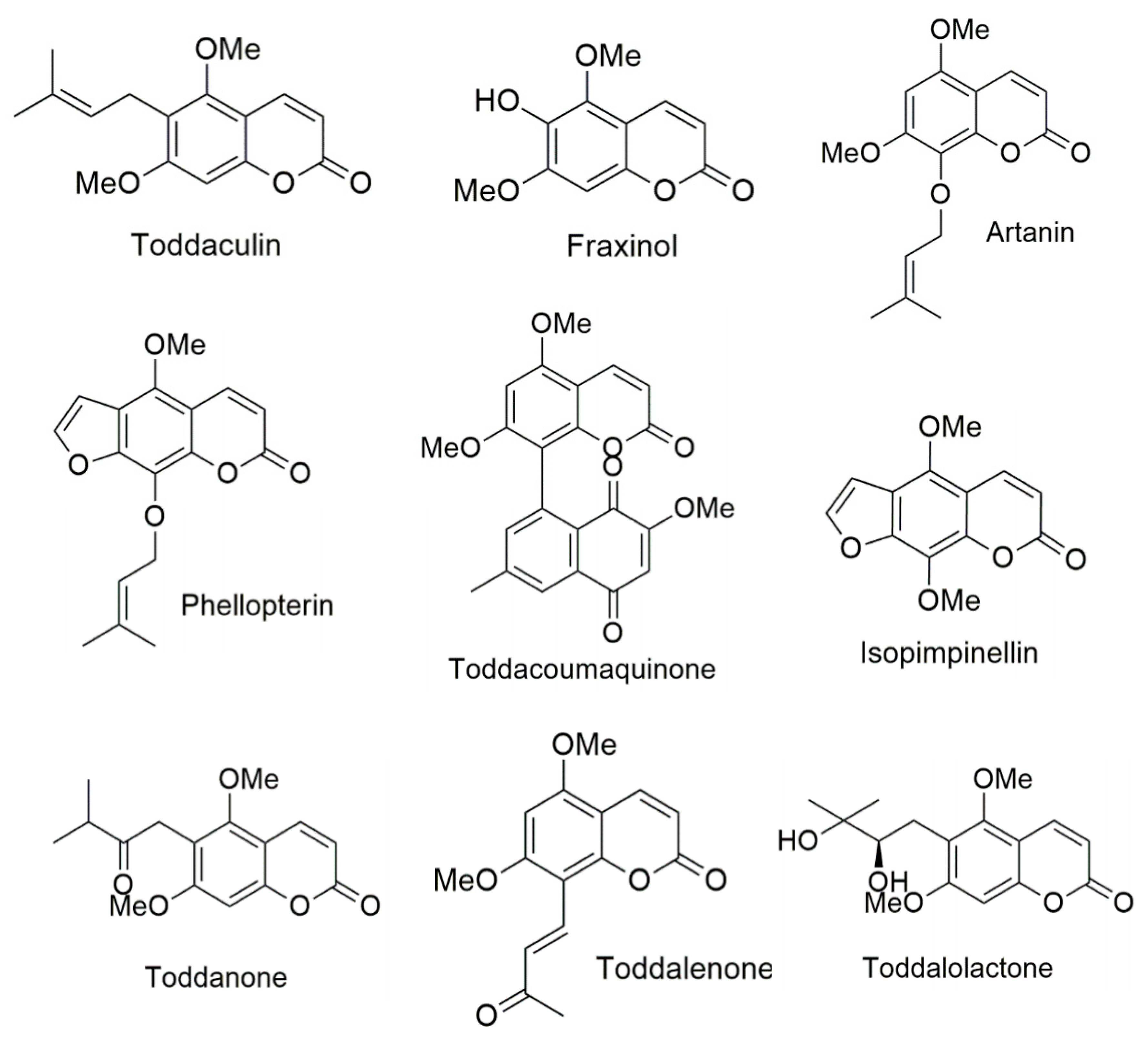
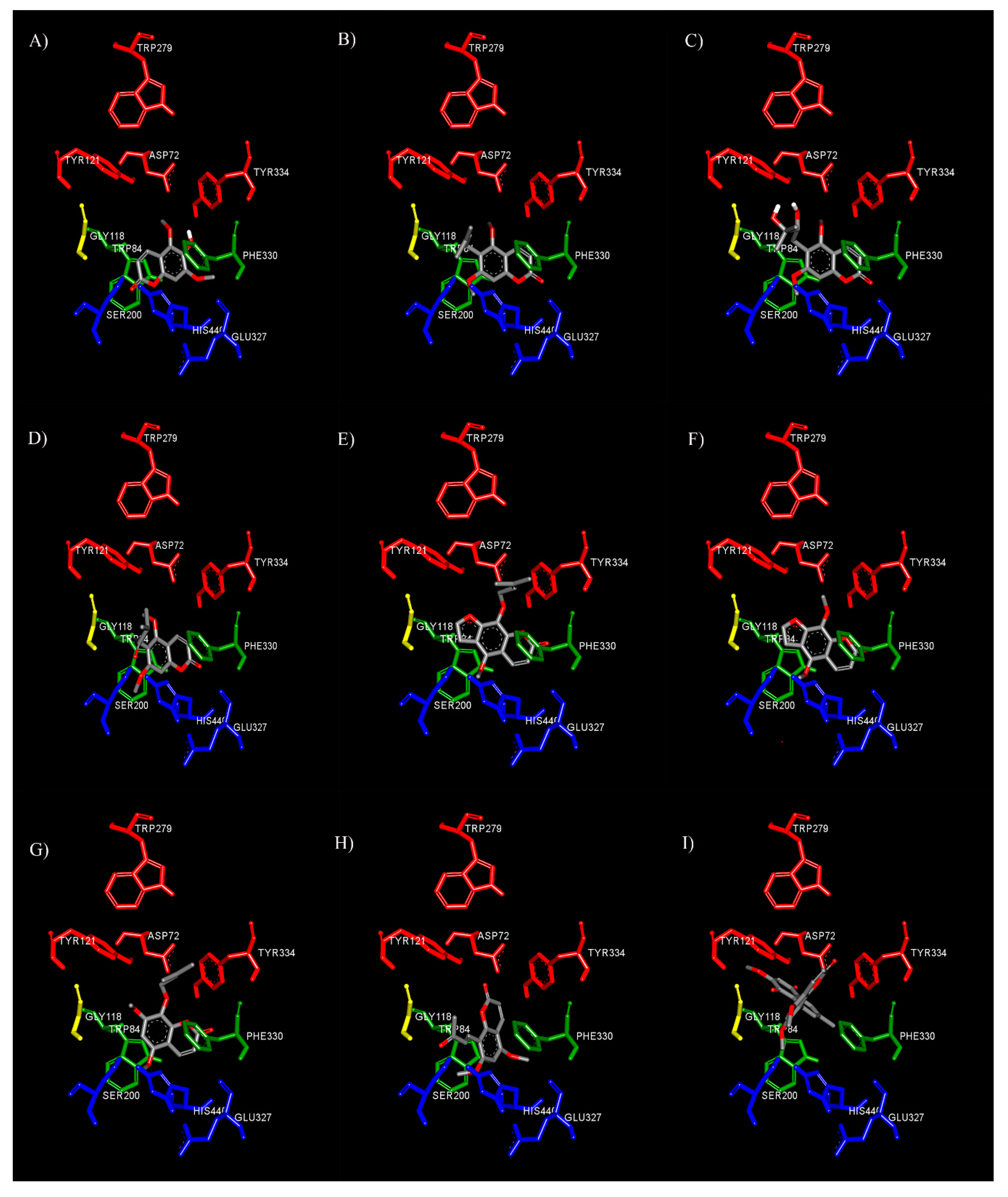
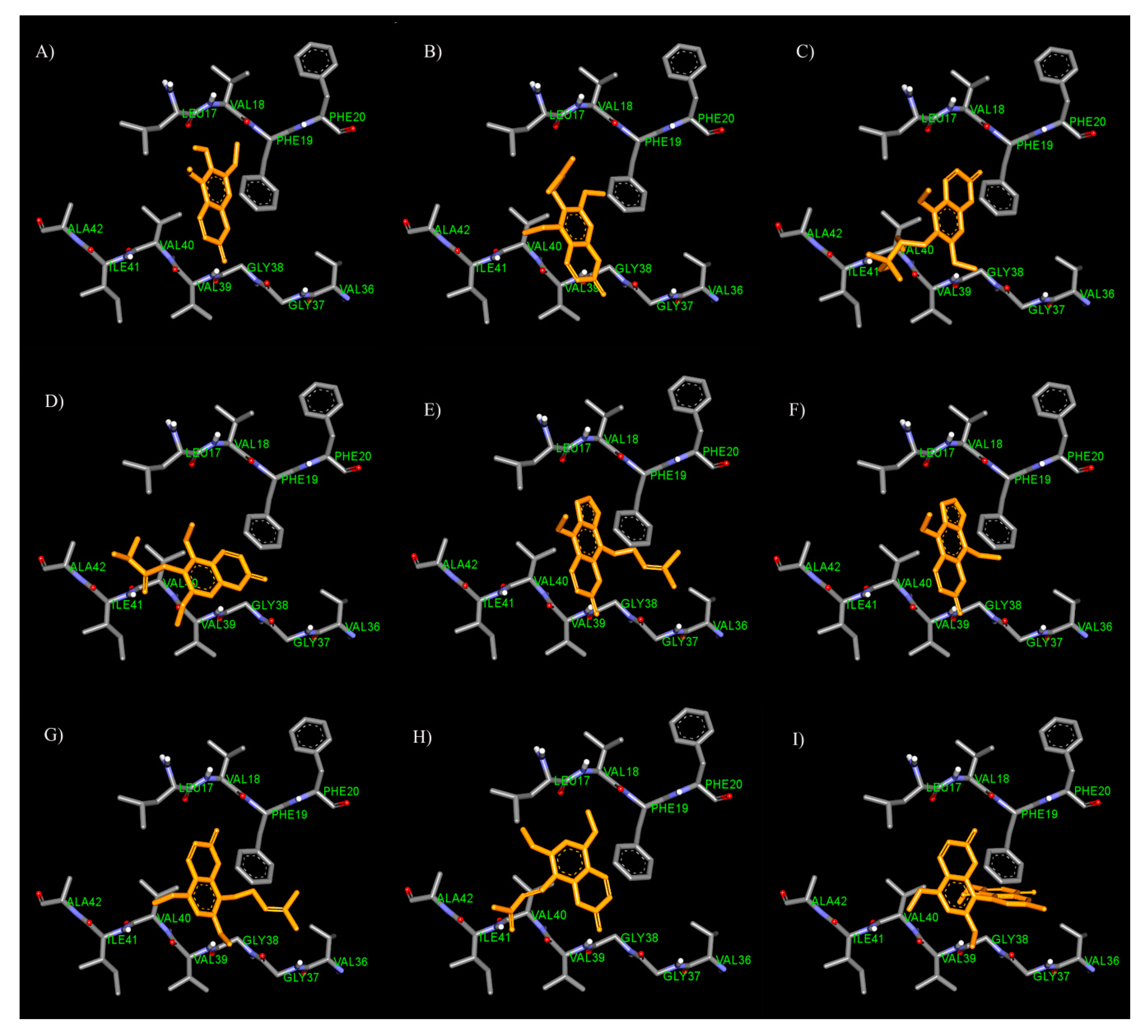
3.4. Binding Interaction Study by Molecular Docking
3.4.1. Binding Interaction with AChE
3.4.2. Binding Interaction with Aβ
3.5. Neuroprotective Activity against H2O2 and Aβ1–42-Induced Neuronal Cell Death
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | amyloid beta |
| AChE | acetylcholinesterase |
| ACh | acetylcholine |
| CAS | catalytic anionic site |
| PAS | peripheral anionic site |
| APP | amyloid precursor protein |
| MTDLs | multi-target directed ligands |
| ThT | Thioflavin T |
| SAR | the structure–activity relationship |
| H2O2 | hydrogen peroxide |
| MTT | 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide |
| DTNB | 5,5′-dithiobis(2-nitrobenzoic acid) |
| DMSO | dimethyl sulfoxide |
References
- Jahn, H. Memory loss in Alzheimer’s disease. Dialogues Clin. Neurosci. 2013, 15, 445–454. [Google Scholar] [PubMed]
- Tarawneh, R.; Holtzman, D.M. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb. Perspect. Med. 2012, 2, 1–16. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; LeVine, H. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeo, J. The role of oxidative stress in Alzheimer ’ s Disease. J. Alzheimer’s Dis. Park. 2013, 03. [Google Scholar] [CrossRef]
- Hasselmo, M.E. The role of acetylcholine in learning and memory. Curr. Opin. Neurobiol. 2006, 16, 710–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [Green Version]
- Pinho, B.R.; Ferreres, F.; Valentão, P.; Andrade, P.B. Nature as a source of metabolites with cholinesterase-inhibitory activity: An approach to Alzheimer’s disease treatment. J. Pharm. Pharmacol. 2013, 65, 1681–1700. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; Canales, M.A.; Shin, I.; Weiner, L.M.; Silman, I.; Inestrosa, N.C. A structural motif of acetylcholinesterase that promotes amyloid β-peptide fibril formation. Biochemistry 2001, 40, 10447–10457. [Google Scholar] [CrossRef]
- Kwon, Y.E.; Park, J.Y.; No, K.T.; Shin, J.H.; Lee, S.K.; Eun, J.S.; Yang, J.H.; Shin, T.Y.; Kim, D.K.; Chae, B.S.; et al. Synthesis, in vitro assay, and molecular modeling of new piperidine derivatives having dual inhibitory potency against acetylcholinesterase and Abeta1-42 aggregation for Alzheimer’s disease therapeutics. Bioorg. Med. Chem. 2007, 15, 6596–6607. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Alvarez, A.; Pérez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer’s fibrils: Possible role of the peripheral site of the enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Madav, Y.; Wairkar, S.; Prabhakar, B. Recent therapeutic strategies targeting beta amyloid and tauopathies in Alzheimer’s disease. Brain Res. Bull. 2019, 146, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Singh, B.; Singh, N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorg. Med. Chem. 2012, 20, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Jannat, S.; Jung, H.A.; Choi, R.J.; Roy, A.; Choi, J.S. Anti-Alzheimer’s disease potential of coumarins from Angelica decursiva and Artemisia capillaris and structure-activity analysis. Asian Pac. J. Trop. Med. 2016, 9, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Thiratmatrakul, S.; Yenjai, C.; Waiwut, P.; Vajragupta, O.; Reubroycharoen, P.; Tohda, M.; Boonyarat, C. Synthesis, biological evaluation and molecular modeling study of novel tacrine-carbazole hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014, 75, 21–30. [Google Scholar] [CrossRef]
- Zhao, Q.; Brett, M.; Van Osselaer, N.; Huang, F.; Raoult, A.; Van Peer, A.; Verhaeghe, T.; Hust, R. Galantamine pharmacokinetics, safety, and tolerability profiles are similar in healthy Caucasian and Japanese subjects. J. Clin. Pharmacol. 2002, 42, 1002–1010. [Google Scholar] [CrossRef]
- Ji, H.; Zhang, H. Multipotent natural agents to combat Alzheimer’s disease. Functional spectrum and structural features. Acta Pharmacol. Sin. 2008, 29, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, M.; Chandra, R.H.; Asres, K.; Veeresham, C. Toddalia asiatica (Linn.) Lam.—A Comprehensive Review. Pharmacogn. Rev. 2008, 2, 386–397. [Google Scholar]
- Orwa, J.A.; Jondiko, I.J.O.; Minja, R.J.A.; Bekunda, M. The use of Toddalia asiatica (L) Lam. (Rutaceae) in traditional medicine practice in East Africa. J. Ethnopharmacol. 2008, 115, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Orwa, J.A.; Ngeny, L.; Mwikwabe, N.M.; Ondicho, J.; Jondiko, I.J.O. Antimalarial and safety evaluation of extracts from Toddalia asiatica (L) Lam. (Rutaceae). J. Ethnopharmacol. 2013, 145, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Piller, N.B. A comparison of the effectiveness of some anti-inflammatory drugs on thermal oedema. Br. J. Exp. Pathol. 1975, 56, 554–560. [Google Scholar]
- Kumagai, M.; Watanabe, A.; Yoshida, I.; Mishima, T.; Nakamura, M.; Nishikawa, K.; Morimoto, Y. Evaluation of aculeatin and toddaculin isolated from Toddalia asiatica as anti-inflammatory agents in LPS-stimulated RAW264 macrophages. Biol. Pharm. Bull. 2018, 41, 132–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez, R.; Riveiro, M.E.; Vermeulen, M.; Mondillo, C.; Coombes, P.H.; Crouch, N.R.; Ismail, F.; Mulholland, D.A.; Baldi, A.; Shayo, C.; et al. Toddaculin, a natural coumarin from Toddalia asiatica, induces differentiation and apoptosis in U-937 leukemic cells. Phytomedicine 2012, 19, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, T.; Tahara, S.; Takabayashi, F. Suppression of lipid hydroperoxide-induced oxidative damage to cellular DNA by esculetin. Biol. Pharm. Bull. 2003, 26, 840–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, G.A.; Connell, W.F. Effect of bishydroxycoumarin (dicumarol) on clotting time of whole blood. J. Am. Med. Assoc. 1956, 161, 806. [Google Scholar] [CrossRef]
- Basile, A.; Sorbo, S.; Spadaro, V.; Bruno, M.; Maggio, A.; Faraone, N.; Rosselli, S. Antimicrobial and antioxidant activities of coumarins from the roots of Ferulago campestris (Apiaceae). Molecules 2009, 14, 939–952. [Google Scholar] [CrossRef] [Green Version]
- Abu-Aisheh, M.N.; Al-Aboudi, A.; Mustafa, M.S.; El-Abadelah, M.M.; Ali, S.Y.; Ul-Haq, Z.; Mubarak, M.S. Coumarin derivatives as acetyl- and butyrylcholinestrase inhibitors: An in vitro, molecular docking, and molecular dynamics simulations study. Heliyon 2019, 5, e01552. [Google Scholar] [CrossRef] [Green Version]
- Sukieum, S.; Sang-aroon, W.; Yenjai, C. Coumarins and alkaloids from the roots of Toddalia asiatica. Nat. Prod. Res. 2018, 32, 944–952. [Google Scholar] [CrossRef]
- Hirunwong, C.; Sukieum, S.; Phatchana, R.; Yenjai, C. Cytotoxic and antimalarial constituents from the roots of Toddalia asiatica. Phytochem. Lett. 2016, 17, 242–246. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Bartolini, M.; Bertucci, C.; Cavrini, V.; Andrisano, V. β-Amyloid aggregation induced by human acetylcholinesterase: Inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [Google Scholar] [CrossRef]
- LeVine, H. Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999, 309, 274–284. [Google Scholar]
- Kang, S.Y.; Lee, K.Y.; Sung, S.H.; Park, M.J.; Kim, Y.C. Coumarins isolated from Angelica gigas inhibit Acetylcholinesterase: Structure−Activity Relationships. J. Nat. Prod. 2001, 64, 683–685. [Google Scholar] [CrossRef]
- Fallarero, A.; Oinonen, P.; Gupta, S.; Blom, P.; Galkin, A.; Mohan, C.G.; Vuorela, P.M. Inhibition of acetylcholinesterase by coumarins: The case of coumarin 106. Pharmacol. Res. 2008, 58, 215–221. [Google Scholar] [CrossRef]
- Muñoz, F.J.; Inestrosa, N.C. Neurotoxicity of acetylcholinesterase amyloid β-peptide aggregates is dependent on the type of Aβ peptide and the AChE concentration present in the complexes. FEBS Lett. 1999, 450, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Roychaudhuri, R.; Yang, M.; Hoshi, M.M.; Teplow, D.B. Amyloid β-protein assembly and Alzheimer disease. J. Biol. Chem. 2009, 284, 4749–4753. [Google Scholar] [CrossRef] [Green Version]
- Luhrs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Dobeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid- (1-42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [Green Version]
- Bitan, G.; Kirkitadze, M.D.; Lomakin, A.; Vollers, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid beta -protein (Abeta) assembly: Abeta 40 and Abeta 42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R. A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 16742–16747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cukalevski, R.; Boland, B.; Frohm, B.; Thulin, E.; Walsh, D.; Linse, S. Role of aromatic side chains in amyloid β-protein aggregation. ACS Chem. Neurosci. 2012, 3, 1008–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexi, T.; Borlongan, C.V.; Faull, R.L.; Williams, C.E.; Clark, R.G.; Gluckman, P.D.; Hughes, P.E. Neuroprotective strategies for basal ganglia degeneration: Parkinson’s and Huntington’s diseases. Prog. Neurobiol. 2000, 60, 409–470. [Google Scholar] [CrossRef]
- Hampton, M.B.; Orrenius, S. Dual regulation of caspase activity by hydrogen peroxide: Implications for apoptosis. FEBS Lett. 1997, 414, 552–556. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, L.; Yin, J.; Luo, Y.; Huang, S. Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. Int. J. Biochem. Cell Biol. 2009, 41, 1284–1295. [Google Scholar] [CrossRef]
- Sanvicens, N.; Gomez-Vicente, V.; Messeguer, A.; Cotter, T.G. The radical scavenger CR-6 protects SH-SY5Y neuroblastoma cells from oxidative stress-induced apoptosis: Effect on survival pathways. J. Neurochem. 2006, 98, 735–747. [Google Scholar] [CrossRef]
- Yu, Y.; Du, J.-R.; Wang, C.-Y.; Qian, Z.-M. Protection against hydrogen peroxide-induced injury by Z-ligustilide in PC12 cells. Exp. Brain Res. 2008, 184, 307–312. [Google Scholar] [CrossRef]
- Nakajima, Y.; Inokuchi, Y.; Nishi, M.; Shimazawa, M.; Otsubo, K.; Hara, H. Coenzyme Q10 protects retinal cells against oxidative stress in vitro and in vivo. Brain Res. 2008, 1226, 226–233. [Google Scholar] [CrossRef]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-Beta: A crucial factor in Alzheimer’s Disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.; Shakeri, A.; Rao, P.P.N. Amyloid cascade in Alzheimer’s disease: Recent advances in medicinal chemistry. Eur. J. Med. Chem. 2016, 113, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Mora, P.; Luna, R.; Colín-Barenque, L. Amyloid beta: Multiple mechanisms of toxicity and only some protective effects? Oxid. Med. Cell. Longev. 2014, 2014, 795375. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, Y.; Yan, J.; Zhao, X.; Sun, X.; Zhang, Y.; Guo, J.; Zhu, C. Acteoside protects human neuroblastoma SH-SY5Y cells against β-amyloid-induced cell injury. Brain Res. 2009, 1283, 139–147. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | AChE assay IC50 (μM) | AChE-Induce Aβ IC50 (μM) | Self-Induced Aβ IC50 (μM) |
|---|---|---|---|
| Phellopterin | 38 ± 3 | 97 ± 7 | 95 ± 3 |
| Isopimpinellin | 41 ± 1 | IC50 > 500 μM | 125 ± 13 |
| Toddalolactone | 49 ± 5 | 105 ± 9 | 220 ± 1 |
| Toddacoumaquinone | 46 ± 12 | 72 ± 5 | 145 ± 2 |
| Toddanone | 46 ± 3 | 110 ± 2 | 76 ± 1 |
| Toddaculin | 53 ± 9 | 232 ± 10 | 143 ± 4 |
| Toddalenone | 40 ± 4 | 211 ± 29 | 99 ± 1 |
| Artanin | 51 ± 2 | 98 ± 19 | 124 ± 10 |
| Fraxinol | 18 ± 7 | IC50 > 500 μM | 209 ± 21 |
| Tacrine | 0.28 ± 0.04 | Not detected | Not detected |
| Curcumin | Not detected | 5.8 ± 0.3 | 5.0 ± 0.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takomthong, P.; Waiwut, P.; Yenjai, C.; Sripanidkulchai, B.; Reubroycharoen, P.; Lai, R.; Kamau, P.; Boonyarat, C. Structure–Activity Analysis and Molecular Docking Studies of Coumarins from Toddalia asiatica as Multifunctional Agents for Alzheimer’s Disease. Biomedicines 2020, 8, 107. https://doi.org/10.3390/biomedicines8050107
Takomthong P, Waiwut P, Yenjai C, Sripanidkulchai B, Reubroycharoen P, Lai R, Kamau P, Boonyarat C. Structure–Activity Analysis and Molecular Docking Studies of Coumarins from Toddalia asiatica as Multifunctional Agents for Alzheimer’s Disease. Biomedicines. 2020; 8(5):107. https://doi.org/10.3390/biomedicines8050107
Chicago/Turabian StyleTakomthong, Pitchayakarn, Pornthip Waiwut, Chavi Yenjai, Bungon Sripanidkulchai, Prasert Reubroycharoen, Ren Lai, Peter Kamau, and Chantana Boonyarat. 2020. "Structure–Activity Analysis and Molecular Docking Studies of Coumarins from Toddalia asiatica as Multifunctional Agents for Alzheimer’s Disease" Biomedicines 8, no. 5: 107. https://doi.org/10.3390/biomedicines8050107