A Non-Coding RNA Landscape of Bronchial Epitheliums of Lung Cancer Patients

 , ,
, ,
Abstract
:1. Introduction
2. Experimental Section
2.1. Collecting Sputum and Enriching Exfoliated Bronchial Epithelial Cells
2.2. NGS Analysis
2.3. Reverse Transcription-PCR (RT-PCR) Analysis of NcRNAs in Sputum
2.4. RNA Interference
2.5. Cell Viability, Proliferation, and Colony Formation Assays
2.6. Transmigration and Wound Healing Assays
2.7. Cell Cycle Analysis by Flow Cytometry
2.8. Tumorigenicity Assays in Nude Mice
2.9. Statistical Analysis
3. Results
3.1. Study Population
3.2. Enriched Bronchial Epithelial Cells of Sputum
3.3. Differentially Expressed ncRNAs in Bronchial Epithelial Cells from Sputum of Stage I NSCLC Patients versus Cancer-Free Smokers
3.4. Validation of the ncRNAs in Sputum of Stage I NSCLC Patients versus Cancer-Free Smokers
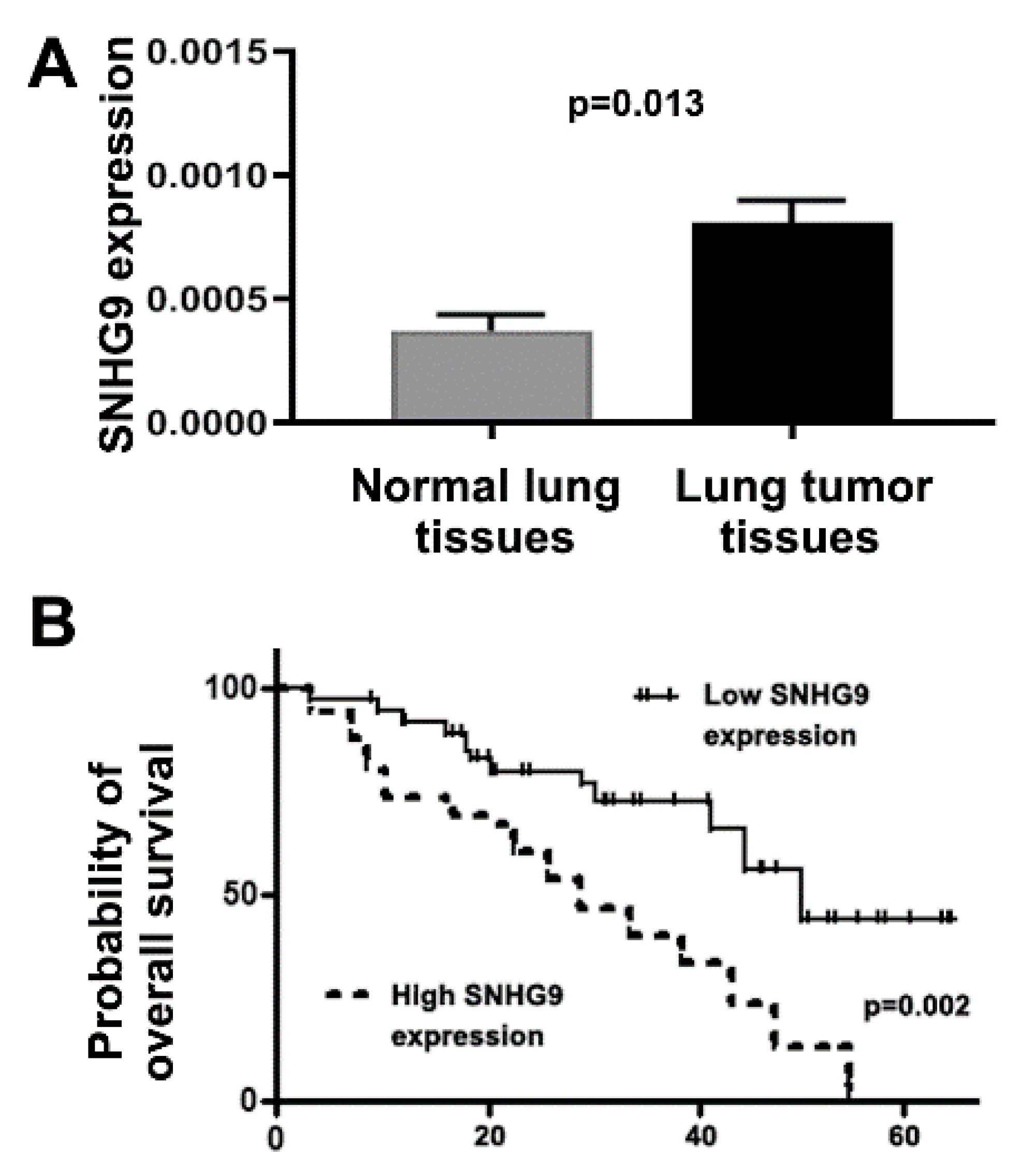
3.5. Dysregulation of SNHG9 Contributes to Tumorigenesis of NSCLC
3.6. Upregulation of SNHG9 Is Associated with Advanced Stage and Survival of Lung Cancer Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2020. [Google Scholar]
- Tang, X.; Shigematsu, H.; Bekele, B.N.; Roth, J.A.; Minna, J.D.; Hong, W.K.; Gazdar, A.F.; Wistuba, I.I. EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res. 2005, 65, 7568–7572. [Google Scholar] [CrossRef] [Green Version]
- Romano, G.; Veneziano, D.; Acunzo, M.; Croce, C.M. Small non-coding RNA and cancer. Carcinogenesis 2017, 38, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Sozzi, G.; Pastorino, U.; Croce, C.M. MicroRNAs and lung cancer: From markers to targets. Cell Cycle 2011, 10, 2045–2046. [Google Scholar] [CrossRef] [Green Version]
- Irimie, A.I.; Zimta, A.A.; Ciocan, C.; Mehterov, N.; Dudea, D.; Braicu, C.; Berindan-Neagoe, I. The Unforeseen Non-Coding RNAs in Head and Neck Cancer. Genes 2018, 9, 134. [Google Scholar] [CrossRef] [Green Version]
- Pang, W.; Tian, X.; Bai, F.; Han, R.; Wang, J.; Shen, H.; Zhang, X.; Liu, Y.; Yan, X.; Jiang, F.; et al. Pim-1 kinase is a target of miR-486-5p and eukaryotic translation initiation factor 4E, and plays a critical role in lung cancer. Mol. Cancer 2014, 13, 240. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Tian, X.; Han, R.; Zhang, X.; Wang, X.; Shen, H.; Xue, L.; Liu, Y.; Yan, X.; Shen, J.; et al. Downregulation of miR-486-5p contributes to tumor progression and metastasis by targeting protumorigenic ARHGAP5 in lung cancer. Oncogene 2014, 33, 1181–1189. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Liao, J.; Guarnera, M.A.; Fang, H.; Cai, L.; Stass, S.A.; Jiang, F. Analysis of MicroRNAs in sputum to improve computed tomography for lung cancer diagnosis. J. Thorac. Oncol. 2014, 9, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Todd, N.W.; Liu, Z.; Zhan, M.; Fang, H.; Peng, H.; Alattar, M.; Deepak, J.; Stass, S.A.; Jiang, F. Altered miRNA expression in sputum for diagnosis of non-small cell lung cancer. Lung Cancer 2010, 67, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Todd, N.W.; Yu, L.; Fang, H.; Jiang, F. Early detection of squamous cell lung cancer in sputum by a panel of microRNA markers. Mod. Pathol. 2010, 23, 1157–1164. [Google Scholar] [CrossRef]
- Yu, L.; Todd, N.W.; Xing, L.; Xie, Y.; Zhang, H.; Liu, Z.; Fang, H.; Zhang, J.; Katz, R.L.; Jiang, F. Early detection of lung adenocarcinoma in sputum by a panel of microRNA markers. Int. J. Cancer 2010, 127, 2870–2878. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Su, J.; Guarnera, M.A.; Zhang, H.; Cai, L.; Zhou, R.; Stass, S.A.; Jiang, F. Sputum microRNA biomarkers for identifying lung cancer in indeterminate solitary pulmonary nodules. Clin. Cancer Res. 2015, 21, 484–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.; Liao, J.; Gao, L.; Shen, J.; Guarnera, M.A.; Zhan, M.; Fang, H.; Stass, S.A.; Jiang, F. Analysis of small nucleolar RNAs in sputum for lung cancer diagnosis. Oncotarget 2016, 7, 5131–5142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Guarnera, M.A.; Fang, H.; Jiang, F. Small non-coding RNA biomarkers in sputum for lung cancer diagnosis. Mol. Cancer 2016, 15, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Shen, J.; Mannoor, K.; Guarnera, M.; Jiang, F. Identification of ENO1 As a Potential Sputum Biomarker for Early-Stage Lung Cancer by Shotgun Proteomics. Clin. Lung Cancer 2014, 15, 372–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Ma, J.; Guarnera, M.A.; Fang, H.; Cai, L.; Jiang, F. Digital PCR quantification of miRNAs in sputum for diagnosis of lung cancer. J. Cancer Res. Clin. Oncol. 2014, 140, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Anjuman, N.; Li, N.; Guarnera, M.; Stass, S.A.; Jiang, F. Evaluation of lung flute in sputum samples for molecular analysis of lung cancer. Clin. Transl. Med. 2013, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Todd, N.W.; Li, R.; Zhang, H.; Fang, H.; Stass, S.A. A panel of sputum-based genomic marker for early detection of lung cancer. Cancer Prev. Res. (Phila) 2010, 3, 1571–1578. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Todd, N.W.; Qiu, Q.; Liu, Z.; Katz, R.L.; Stass, S.A. Combined genetic analysis of sputum and computed tomography for noninvasive diagnosis of non-small-cell lung cancer. Lung Cancer 2009, 66, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Q.; Todd, N.W.; Li, R.; Peng, H.; Liu, Z.; Yfantis, H.G.; Katz, R.L.; Stass, S.A.; Jiang, F. Magnetic enrichment of bronchial epithelial cells from sputum for lung cancer diagnosis. Cancer 2008, 114, 275–283. [Google Scholar] [CrossRef]
- Li, R.; Todd, N.W.; Qiu, Q.; Fan, T.; Zhao, R.Y.; Rodgers, W.H.; Fang, H.-B.; Katz, R.L.; Stass, S.A.; Jiang, F. Genetic deletions in sputum as diagnostic markers for early detection of stage I non-small cell lung cancer. Clin. Cancer Res. 2007, 13 Pt 1, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Stass, S.A.; Jiang, F. MicroRNAs as potential biomarkers in human solid tumors. Cancer Lett. 2013, 329, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Jiang, F. Applications of MicroRNAs in the Diagnosis and Prognosis of Lung Cancer. Expert Opin. Med. Diagn. 2012, 6, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Liu, Z.; Todd, N.W.; Zhang, H.; Liao, J.; Yu, L.; Guarnera, M.; Li, R.; Cai, L.; Zhan, M.; et al. Diagnosis of lung cancer in individuals with solitary pulmonary nodules by plasma microRNA biomarkers. BMC Cancer 2011, 11, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Todd, N.W.; Zhang, H.; Yu, L.; Lingxiao, X.; Mei, Y.; Guarnera, M.; Liao, J.; Chou, A.; Lu, C.L.; et al. Plasma microRNAs as potential biomarkers for non-small-cell lung cancer. Lab. Investig. 2011, 91, 579–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Mannoor, K.; Gao, L.; Tan, A.; Guarnera, M.A.; Zhan, M.; Shetty, A.; Stass, S.A.; Xing, L.; Jiang, F. Characterization of microRNA transcriptome in lung cancer by next-generation deep sequencing. Mol. Oncol. 2014, 8, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Ma, J.; Mannoor, K.; Guarnera, M.A.; Shetty, A.C.; Zhan, M.; Xing, L.; Stass, S.A.; Jiang, F. Genome-wide small nucleolar RNA expression analysis of lung cancer by next-generation deep sequencing. Int. J. Cancer 2015, 136, E623–E629. [Google Scholar] [CrossRef]
- Lin, Y.; Leng, Q.; Zhan, M.; Jiang, F. A Plasma Long Noncoding RNA Signature for Early Detection of Lung Cancer. Transl. Oncol. 2018, 11, 1225–1231. [Google Scholar] [CrossRef]
- Leng, Q.; Lin, Y.; Zhan, M.; Jiang, F. An integromic signature for lung cancer early detection. Oncotarget 2018, 9, 24684–24692. [Google Scholar] [CrossRef]
- Mei, Y.-P.; Liao, J.-P.; Shen, J.; Yu, L.; Liu, B.; Liu, L.; Li, R.-Y.; Ji, L.; Dorsey, S.G.; Jiang, Z.-R.; et al. Small nucleolar RNA 42 acts as an oncogene in lung tumorigenesis. Oncogene 2012, 31, 2794–2804. [Google Scholar] [CrossRef]
- Mannoor, K.; Shen, J.; Liao, J.; Liu, Z.; Jiang, F. Small nucleolar RNA signatures of lung tumor-initiating cells. Mol. Cancer 2014, 13, 104. [Google Scholar] [CrossRef] [Green Version]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays; Eli Lilly & Company and the National Center for Advancing Translational Sciences: San Antonio, TX, USA, 2004. [Google Scholar]
- Liu, Z.; Jiang, F. Gene identification and survival prediction with Lp Cox regression and novel similarity measure. Int. J. Data Min. Bioinform. 2009, 3, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tan, M.; Jiang, F. Regularized F-measure maximization for feature selection and classification. J. Biomed. Biotechnol. 2009, 2009, 617946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Gartenhaus, R.B.; Tan, M.; Jiang, F.; Jiao, X. Gene and pathway identification with Lp penalized Bayesian logistic regression. BMC Bioinform. 2008, 9, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Jiang, F.; Tian, G.; Wang, S.; Sato, F.; Meltzer, S.J.; Tan, M. Sparse logistic regression with Lp penalty for biomarker identification. Stat. Appl. Genet. Mol. Biol. 2007, 6, 6. [Google Scholar] [CrossRef]
- Liu, P.; Hwang, J.T. Quick calculation for sample size while controlling false discovery rate with application to microarray analysis. Bioinformatics 2007, 23, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Wang, J.; Ouyang, T.; Lu, N.; Lu, J.; Shen, Y.; Bai, Y.; Xie, X.; Ge, Q.-Y. MiR-486-5p Serves as a Good Biomarker in Nonsmall Cell Lung Cancer and Suppresses Cell Growth With the Involvement of a Target PIK3R1. Front. Genet. 2019, 10, 688. [Google Scholar] [CrossRef] [Green Version]
- Pavel, A.B.; Campbell, J.D.; Liu, G.; Elashoff, D.; Dubinett, S.; Smith, K.; Whitney, D.; Lenburg, M.; Spira, A.; AEGIS Study Team. Alterations in Bronchial Airway miRNA Expression for Lung Cancer Detection. Cancer Prev. Res. (Phila) 2017, 10, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Gao, L.; Che, T.; Du, W.; Li, Q.; Gao, Q.; Wang, Z. The effects of 12C6+ irradiation on cell cycle, apoptosis, and expression of caspase-3 in the human lung cancer cell line h1299. Cancer Biother. Radiopharm. 2012, 27, 113–118. [Google Scholar] [CrossRef]
- Weinstein, L.B.; Steitz, J.A. Guided tours: From precursor snoRNA to functional snoRNP. Curr. Opin. Cell Biol. 1999, 11, 378–384. [Google Scholar] [CrossRef]
- Li, D.; Luo, Y.; Gao, Y.; Yang, Y.; Wang, Y.; Xu, Y.; Tan, S.; Zhang, Y.; Duan, J.; Yang, Y. piR-651 promotes tumor formation in non-small cell lung carcinoma through the upregulation of cyclin D1 and CDK4. Int. J. Mol. Med. 2016, 38, 927–936. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Wang, Y.W.; Fang, B.B.; Zhang, S.J.; Cheng, B.L. piR-651 and its function in 95-D lung cancer cells. Biomed. Rep. 2016, 4, 546–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Yao, J.; Shen, B.; Li, G.; Kong, S.; Bi, D.; Pan, S.; Cheng, B. Role of piwi-interacting RNA-651 in the carcinogenesis of non-small cell lung cancer. Oncol. Lett. 2018, 15, 940–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafontaine, D.L.; Tollervey, D. The function and synthesis of ribosomes. Nat. Rev. Mol. Cell Biol. 2001, 2, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, Y.; Lv, Q.; Zhang, J.; Wang, Q.; Gao, F.; Hou, H.; Zhang, H.; Zhang, W.; Li, L. Overexpression of Ribosomal RNA in the Development of Human Cervical Cancer Is Associated with rDNA Promoter Hypomethylation. PLoS ONE 2016, 11, e0163340. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, H.; Lam, K.C.; Dong, Y.; Zhang, X.; Lee, C.K.; Zhang, J.; Ng, S.C.; Ng, S.S.M.; Zheng, S.; Chen, Y.; et al. Pre-45s rRNA promotes colon cancer and is associated with poor survival of CRC patients. Oncogene 2017, 36, 6109–6118. [Google Scholar] [CrossRef] [Green Version]
- Randerath, K.; Agrawal, H.P.; Randerath, E. tRNA alterations in cancer. Recent Results Cancer Res. 1983, 84, 103–120. [Google Scholar]
- Shao, Y.; Sun, Q.; Liu, X.; Wang, P.; Wu, R.; Ma, Z. tRF-Leu-CAG promotes cell proliferation and cell cycle in non-small cell lung cancer. Chem. Biol. Drug Des. 2017, 90, 730–738. [Google Scholar] [CrossRef]
- Kim, E.Y.; Jung, J.Y.; Kim, A.; Kim, K.; Chang, Y.S. Methionyl-tRNA synthetase overexpression is associated with poor clinical outcomes in non-small cell lung cancer. BMC Cancer 2017, 17, 467. [Google Scholar] [CrossRef]
- Hoskins, A.A.; Moore, M.J. The spliceosome: A flexible, reversible macromolecular machine. Trends Biochem. Sci. 2012, 37, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.M.; Venters, C.C.; Di, C.; Pinto, A.M.; Wan, L.; Younis, I.; Cai, Z.; Arai, C.; So, B.R.; Duan, J.; et al. U1 snRNP regulates cancer cell migration and invasion in vitro. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Jiang, Z.; Wang, S.; Zhao, Y.; Song, X.; Xiao, Y.; Yang, S. Long non-coding small nucleolar RNA host genes in digestive cancers. Cancer Med. 2019, 8, 7693–7704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Liu, S.; Fan, J.; Jin, Y.-H.; Tian, B.; Zheng, X.; Fu, H. Nuclear retention of the lncRNA SNHG1 by doxorubicin attenuates hnRNPC-p53 protein interactions. EMBO Rep. 2017, 18, 536–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Lung Cancer Patients | % | Cancer-Free Controls | % | |
|---|---|---|---|---|
| Parameter | ||||
| Mean of age (years) | 68.2 | 65.6 | ||
| Gender | ||||
| Men | 21 | 65.6 | 22 | 66.7 |
| Women | 11 | 34.4 | 11 | 33.3 |
| Race | ||||
| White American | 23 | 71.9 | 23 | 69.7 |
| African American | 9 | 28.1 | 10 | 30.3 |
| Mean of smoking pack-years | 49.3 | 37.8 | ||
| Stage | All are stage I NSCLC | |||
| Histology | ||||
| Adenocarcinoma | 16 | 50 | ||
| Squamous cell carcinoma | 16 | 50 |
| Characteristics | Number of Patients in Each Category (%) |
|---|---|
| Mean of age (years) at diagnosis | 66.9 |
| Sex | |
| Male | 52 (66.7) |
| Female | 26 (33.3) |
| Race | |
| White | 49 (62.8) |
| Black | 29 (37.2) |
| Smoker | |
| Yes | 70 (89.7) |
| No | 8 (10.3) |
| Tumor histology | |
| Squamous cell carcinoma | 39 (50.0) |
| Adenocarcinoma | 39 (50.0) |
| T stage | |
| I | 30 (38.5) |
| II | 29 (37.2) |
| III–IV | 19 (24.3) |
| Genes | FC | Genes | FC |
|---|---|---|---|
| miRs | piRNAs | ||
| MIR-9-1 | 28.146 | piR-004987 | 5.637 |
| MIR-577 | 21.745 | piR-020809 | 5.058 |
| MIR-410 | 21.007 | piR-023338 | −3.489 |
| MIR-487B | 18.242 | piR-011186 | −4.161 |
| MIR-409 | 14.424 | tRNAs | |
| MIR-338 | −7.723 | TRNAV33P | 16.688 |
| MIR-486 | −8.086 | TRNAE27P | 15.772 |
| MIR-135A1 | −19.011 | TRNAG34P | 2.159 |
| MIR-184 | −28.380 | TRNAG32P | 16.774 |
| snRNAs | TRNAE40P | 2.107 | |
| RNU5E-1 | 32.515 | TRNAK42P | −2.006 |
| U4 | 11.005 | rRNAs | |
| RNU7-1 | 6.874 | RN5-8S5 | 4.936 |
| RNU4ATAC | 6.400 | RN5-8S3 | 2.008 |
| RNU5A-1 | 5.736 | RN5-8S2 | 5.151 |
| snoRNAs | RN5248 | −2.188 | |
| SNORD114-20 | 43.008 | lncRNAs | |
| SNORD113-5 | 36.658 | SNHG9 | 8.474 |
| SNORD114-25 | 31.656 | SNHG2 | 7.568 |
| SNORD114-28 | 30.173 | MEG8 | 7.078 |
| SNORD114-26 | 22.244 | LINC00461 | 7.786 |
| SNORD113-7 | 19.579 | SNHG11 | 6.635 |
| SNORD114-21 | 17.826 | GAS5 | −3.082 |
| SNORD33 | 16.235 | TUG1 | −4.866 |
| SNORD114-23 | 16.090 | PANDAR | −5.099 |
| Expression Levels of the ncRNAs Detected by NGS | ||||
| ncRNAs | Median of RPM in NSCLC Patients | Median of RPM in Controls | Log2 Fold Change (FC) of Patients/Controls | p-Value |
| miRNAs | ||||
| miR-21 | 39789.25 | 578.96 | 6.10 | <0.001 |
| miR-31 | 4800.34 | 973.20 | 2.30 | <0.001 |
| miR-210 | 5935.71 | 226.72 | 4.71 | <0.001 |
| MIR-486 | 140.87 | 37632.26 | –8.06 | |
| snoRNAs | ||||
| snoRD66 | 4822.28 | 151.60 | 4.99 | <0.001 |
| snoRD78 | 3983.48 | 161.80 | 4.62 | <0.001 |
| snoRA42 | 4518.27 | 86.72 | 5.70 | <0.001 |
| lncRNAs | ||||
| SNHG9 | 70896.26 | 198.99 | 8.48 | <0.001 |
| H19 | 6073.51 | 160.89 | 5.24 | <0.001 |
| HOTAIR | 2616.69 | 309.18 | 3.08 | 0.0060 |
| Expression levels of the ncRNAs detected by RT-PCR | ||||
| ncRNAs | Mean of Level in NSCLC Patients | Mean of Level in Controls | p-Value | |
| miRNAs | ||||
| miR-21 | 297.26 | 7.16 | 5.38 | <0.001 |
| miR-31 | 4.89 | 0.75 | 2.70 | <0.001 |
| miR-210 | 178.94 | 12.86 | 3.80 | <0.001 |
| MIR-486 | 19 | 378 | −4.31 | |
| snoRNAs | ||||
| snoRD66 | 1.71 | 0.04 | 5.42 | <0.001 |
| snoRD78 | 1.86 | 0.12 | 3.95 | 0.001 |
| snoRA42 | 2.65 | 0.1 | 4.73 | <0.001 |
| lncRNAs | ||||
| SNHG9 | 16.78 | 0.33 | 5.67 | <0.001 |
| H19 | 9.34 | 0.86 | 3.44 | <0.001 |
| HOTAIR | 3.11 | 0.38 | 3.03 | <0.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.; Holden, V.; Dhilipkannah, P.; Deepak, J.; Todd, N.W.; Jiang, F. A Non-Coding RNA Landscape of Bronchial Epitheliums of Lung Cancer Patients. Biomedicines 2020, 8, 88. https://doi.org/10.3390/biomedicines8040088
Lin Y, Holden V, Dhilipkannah P, Deepak J, Todd NW, Jiang F. A Non-Coding RNA Landscape of Bronchial Epitheliums of Lung Cancer Patients. Biomedicines. 2020; 8(4):88. https://doi.org/10.3390/biomedicines8040088
Chicago/Turabian StyleLin, Yanli, Van Holden, Pushpawallie Dhilipkannah, Janaki Deepak, Nevins W. Todd, and Feng Jiang. 2020. "A Non-Coding RNA Landscape of Bronchial Epitheliums of Lung Cancer Patients" Biomedicines 8, no. 4: 88. https://doi.org/10.3390/biomedicines8040088