Basic Biology of Hypoxic Responses Mediated by the Transcription Factor HIFs and Its Implication for Medicine


Abstract
:1. Introduction
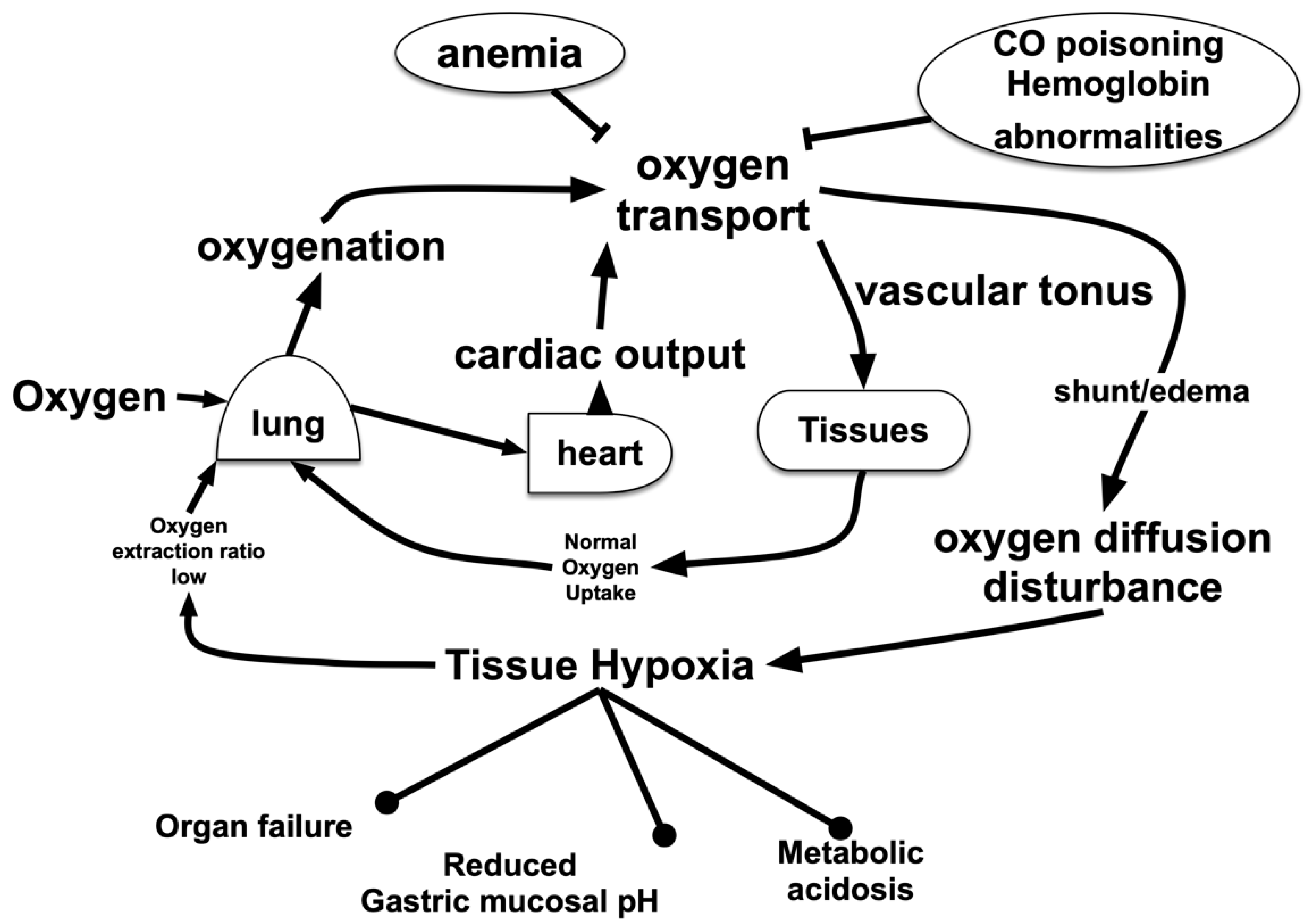
2. Hypoxic Conditions in the Living Body
2.1. Hypoxic Hypoxia
2.2. Hypemic Hypoxia
2.3. Tissue Hypoperfusion and Ischemia
2.4. Tissue Oxygen Metabolism Disorder
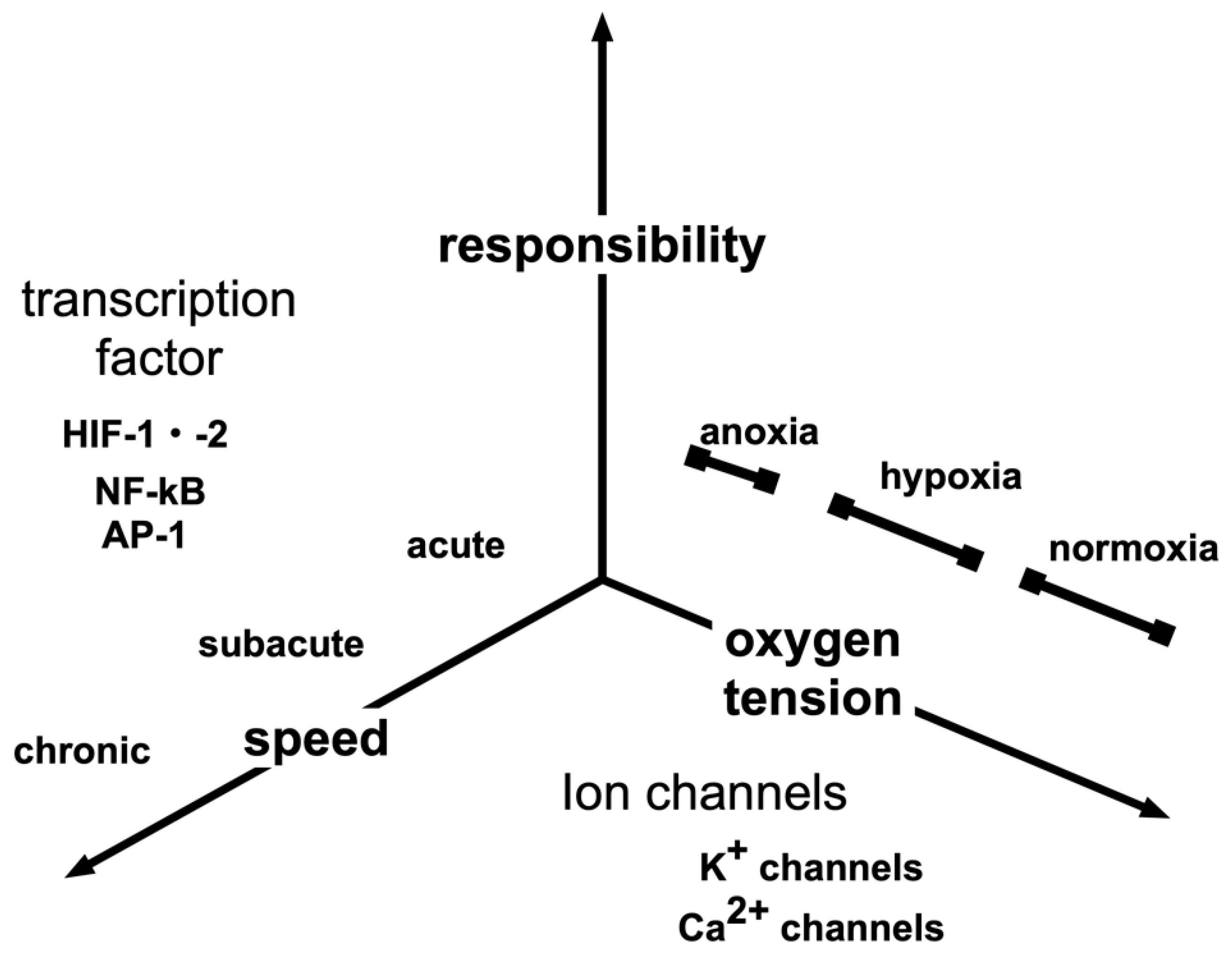
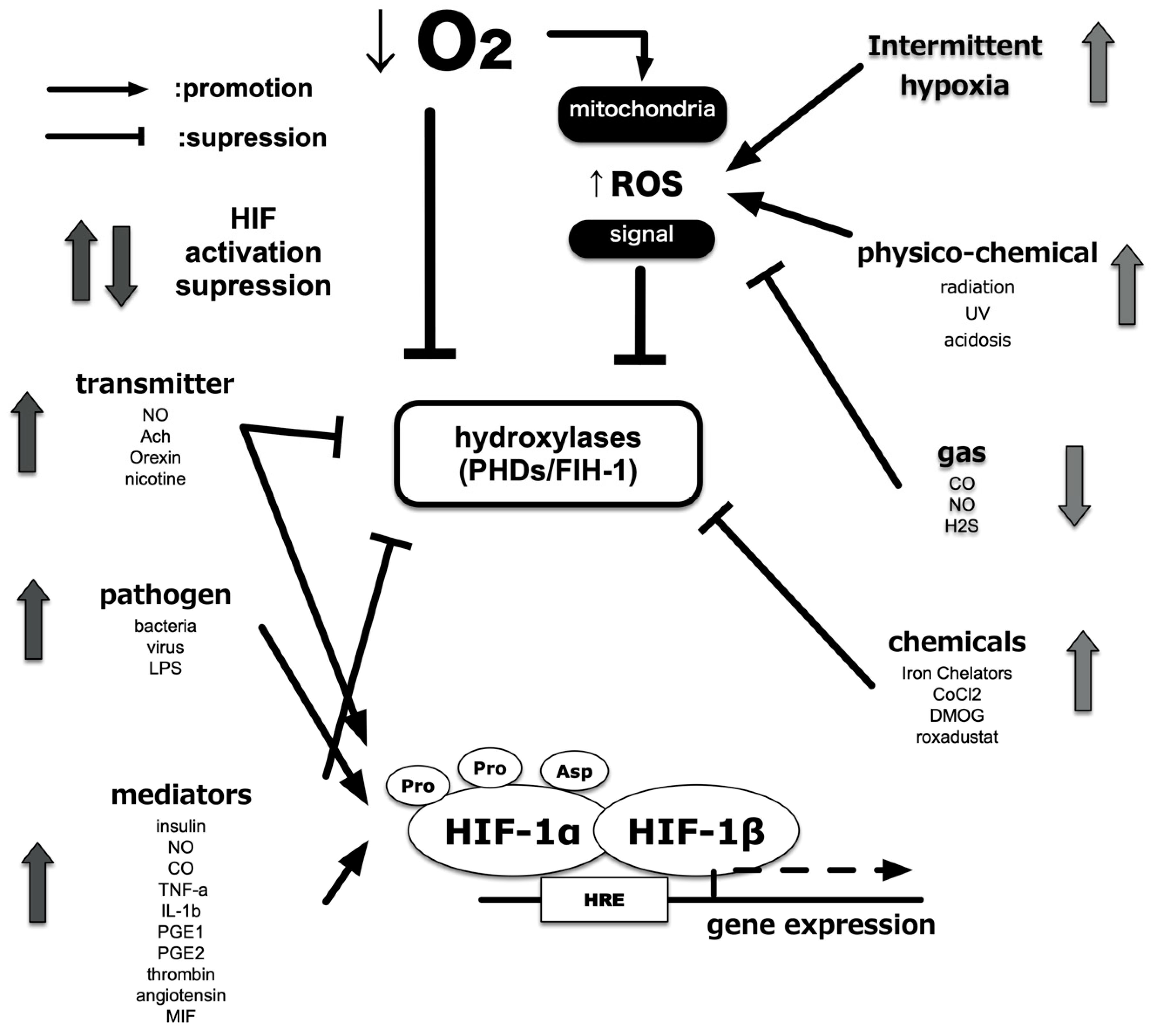
3. Hypoxia-Sensing Mechanisms
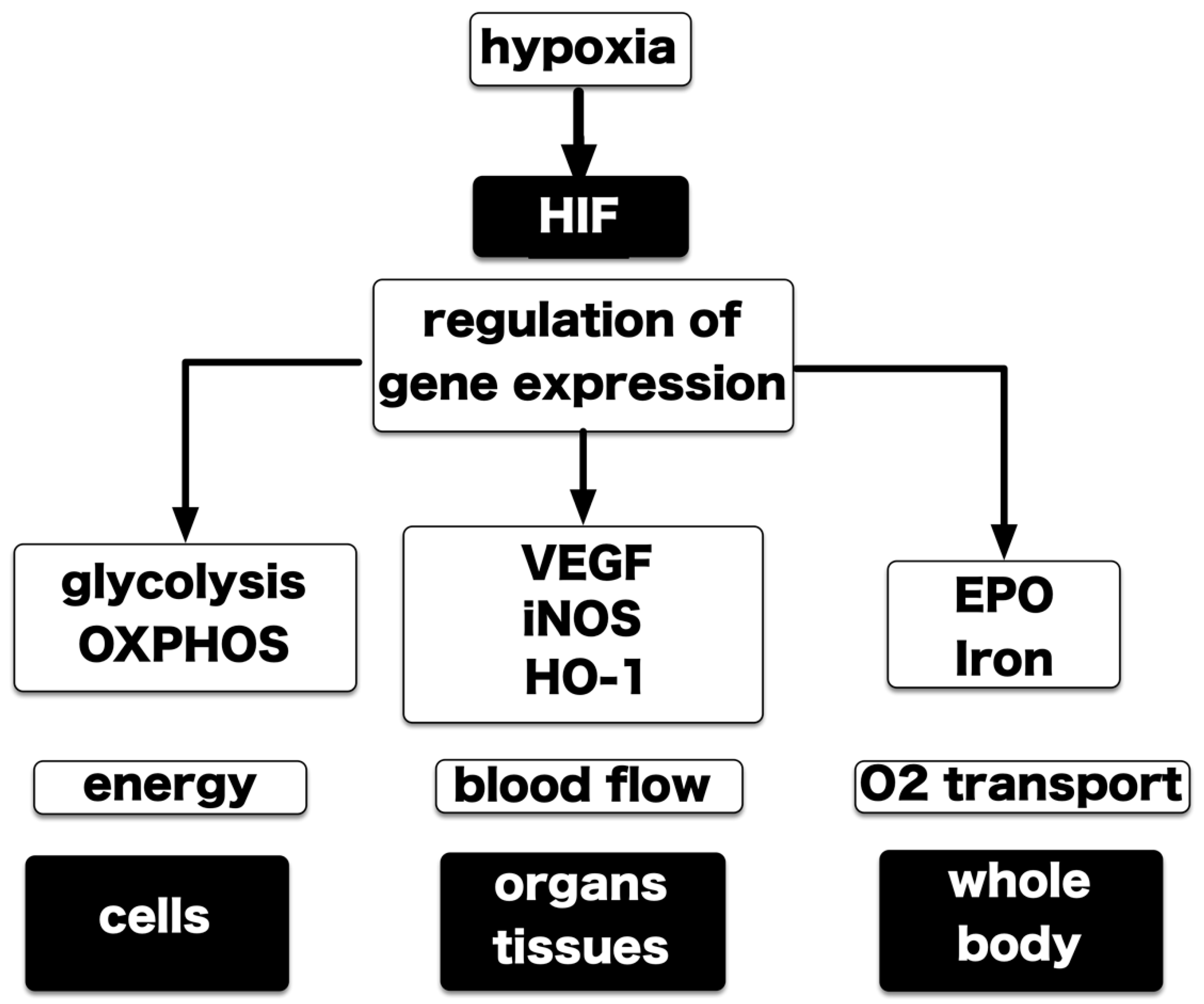
4. Hypoxia-Inducible Factors
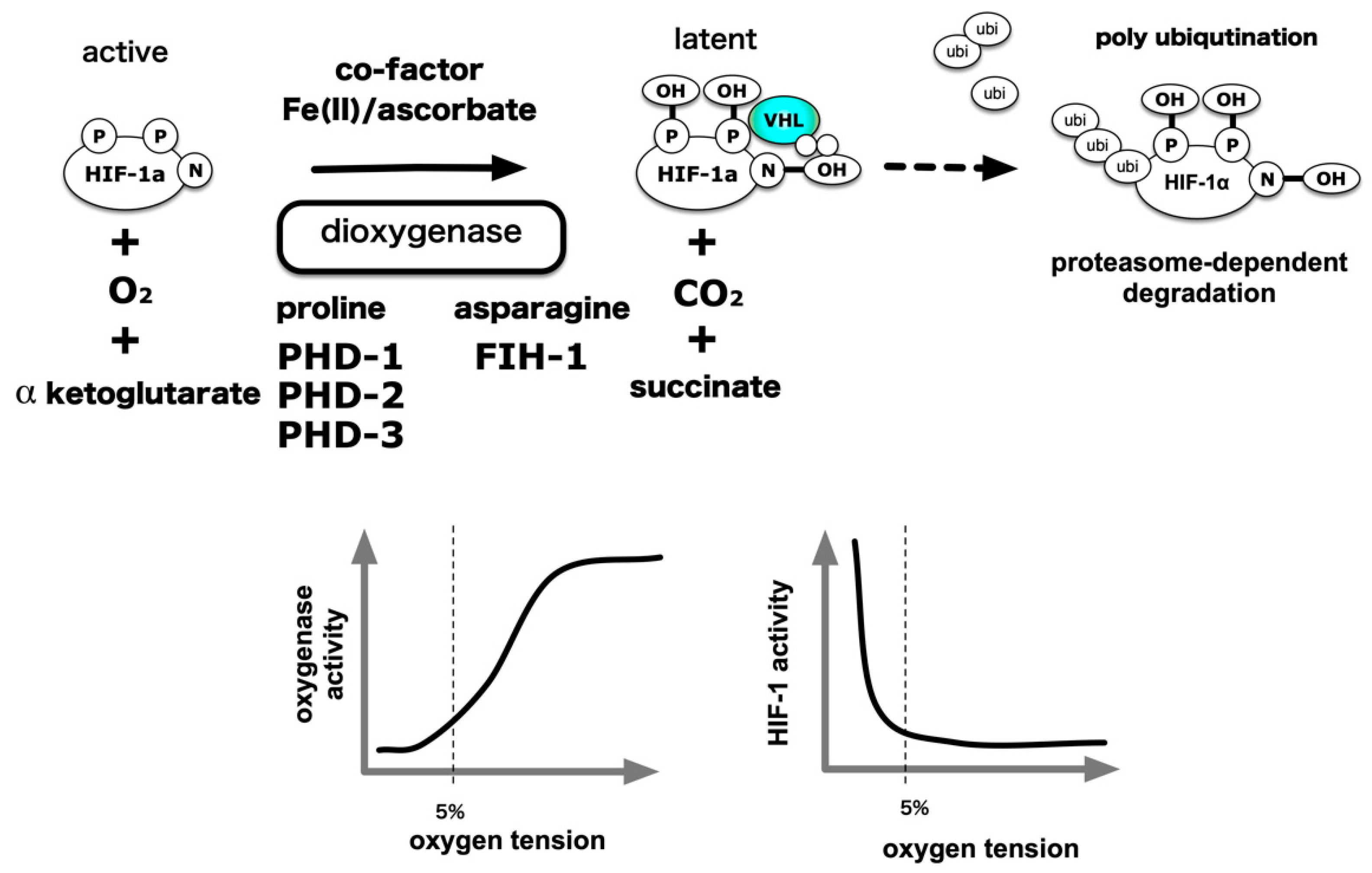
5. Central Dogma of the Molecular Mechanism of HIF-1 Activation
6. ROS Generation under Hypoxic Conditions and Its Involvement in HIF-1 Activation
7. Response to Intermittent Hypoxia
8. Induction of HIF-1 Activity under Non-Hypoxic Conditions
A Close Interaction between NF-κB and HIFs
9. Pathophysiological Role of HIFs in Human Diseases
9.1. Kidney and Iron Metabolism
9.2. Cancer Progression
9.3. Lactate and Hypoxia Response
9.4. Cardiac Hypertrophy and Heart Failure
9.5. Placenta Formation and Oxygen Tension-Pregnancy Induced Hypertension and Intrauterine Growth Retardation
9.6. Immunity
9.7. Role of Hypoxia on Autophagy and Cellular Damage
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Severinghaus, J.W. Eight sages over five centuries share oxygen’s discovery. Adv. Physiol. Educ. 2016, 40, 370–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severinghaus, J.W. The Most Important Discovery of Science. Adv. Exp. Med. Biol. 2016, 876, 1–16. [Google Scholar] [CrossRef] [PubMed]
- West, J.B. Joseph Priestley, oxygen, and the enlightenment. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L111–L119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fara, P. Joseph Priestley: Docter Phlogiston or Reverend Oxygen? Endeavour 2010, 34, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Leach, R.M.; Treacher, D.F. The pulmonary physician in critical care * 2: Oxygen delivery and consumption in the critically ill. Thorax 2002, 57, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, N.T.; Leach, R.M. ABC of oxygen. Acute oxygen therapy. BMJ 1998, 317, 798–801. [Google Scholar] [CrossRef]
- Evans, T.W.; Smithies, M. ABC of intensive care: Organ dysfunction. BMJ 1999, 318, 1606–1609. [Google Scholar] [CrossRef]
- Williams, A.J. ABC of oxygen: Assessing and interpreting arterial blood gases and acid-base balance. BMJ 1998, 317, 1213–1216. [Google Scholar] [CrossRef]
- Grocott, M.P.; Martin, D.S.; Levett, D.Z.; McMorrow, R.; Windsor, J.; Montgomery, H.E.; Caudwell Xtreme Everest Research Group. Arterial blood gases and oxygen content in climbers on Mount Everest. N. Engl. J. Med. 2009, 360, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Griva, K.; Stygall, J.; Wilson, M.H.; Martin, D.; Levett, D.; Mitchell, K.; Mythen, M.; Montgomery, H.E.; Grocott, M.P.; Aref-Adib, G.; et al. Caudwell Xtreme Everest: A prospective study of the effects of environmental hypoxia on cognitive functioning. PLoS ONE 2017, 12, e0174277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, A.; Zibrak, J.D. Carbon monoxide poisoning. N. Engl. J. Med. 1998, 339, 1603–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, J.J.; Wang, L.; Xu, Q.; McTiernan, C.F.; Shiva, S.; Tejero, J.; Gladwin, M.T. Carbon Monoxide Poisoning: Pathogenesis, Management, and Future Directions of Therapy. Am. J. Respir. Crit. Care Med. 2017, 195, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Curry, S. Methemoglobinemia. Ann. Emerg Med. 1982, 11, 214–221. [Google Scholar] [CrossRef]
- Wright, R.O.; Lewander, W.J.; Woolf, A.D. Methemoglobinemia: Etiology, pharmacology, and clinical management. Ann. Emerg. Med. 1999, 34, 646–656. [Google Scholar] [CrossRef]
- Haley, K. Congenital Hemolytic Anemia. Med. Clin. North. Am. 2017, 101, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef]
- Higgs, D.R.; Engel, J.D.; Stamatoyannopoulos, G. Thalassaemia. Lancet 2012, 379, 373–383. [Google Scholar] [CrossRef]
- Tanaka, T.; Kai, S.; Koyama, T.; Daijo, H.; Adachi, T.; Fukuda, K.; Hirota, K. General Anesthetics Inhibit Erythropoietin Induction under Hypoxic Conditions in the Mouse Brain. PLoS ONE 2011, 6, e29378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kai, S.; Tanaka, T.; Matsuyama, T.; Suzuki, K.; Hirota, K. The volatile anesthetic isoflurane differentially suppresses the induction of erythropoietin synthesis elicited by acute anemia and systemic hypoxemia in mice in an hypoxia-inducible factor-2-dependent manner. Eur. J. Pharmacol. 2014, 732C, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Itasaka, S.; Kizaka-Kondoh, S.; Shibuya, K.; Morinibu, A.; Shinomiya, K.; Hiraoka, M. The Akt/mTOR pathway assures the synthesis of HIF-1alpha protein in a glucose- and reoxygenation-dependent manner in irradiated tumors. J. Biol. Chem. 2009, 284, 5332–5342. [Google Scholar] [CrossRef] [Green Version]
- Dayan, F.; Bilton, R.L.; Laferriere, J.; Trottier, E.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Activation of HIF-1alpha in exponentially growing cells via hypoxic stimulation is independent of the Akt/mTOR pathway. J. Cell Physiol. 2009, 218, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Gracia, R.; Shepherd, G. Cyanide poisoning and its treatment. Pharmacotherapy 2004, 24, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Tshala-Katumbay, D.D.; Ngombe, N.N.; Okitundu, D.; David, L.; Westaway, S.K.; Boivin, M.J.; Mumba, N.D.; Banea, J.P. Cyanide and the human brain: Perspectives from a model of food (cassava) poisoning. Ann. N Y Acad. Sci. 2016, 1378, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Lipska, K.J.; Mayo, H.; Bailey, C.J.; McGuire, D.K. Metformin in patients with type 2 diabetes and kidney disease: A systematic review. JAMA 2014, 312, 2668–2675. [Google Scholar] [CrossRef] [Green Version]
- Kreisberg, R.A. Lactate homeostasis and lactic acidosis. Ann. Intern. Med. 1980, 92, 227–237. [Google Scholar] [CrossRef]
- Fudickar, A.; Bein, B. Propofol infusion syndrome: Update of clinical manifestation and pathophysiology. Minerva Anestesiol. 2009, 75, 339–344. [Google Scholar]
- Sumi, C.; Okamoto, A.; Tanaka, H.; Nishi, K.; Kusunoki, M.; Shoji, T.; Uba, T.; Matsuo, Y.; Adachi, T.; Hayashi, J.I.; et al. Propofol induces a metabolic switch to glycolysis and cell death in a mitochondrial electron transport chain-dependent manner. PLoS ONE 2018, 13, e0192796. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.J. Acute oxygen sensing by the carotid body: From mitochondria to plasma membrane. J. Appl. Physiol. (1985) 2017, 123, 1335–1343. [Google Scholar] [CrossRef]
- Piskuric, N.A.; Nurse, C.A. Expanding role of ATP as a versatile messenger at carotid and aortic body chemoreceptors. J. Physiol. 2013, 591, 415–422. [Google Scholar] [CrossRef]
- Conde, S.V.; Sacramento, J.F.; Guarino, M.P. Carotid body: A metabolic sensor implicated in insulin resistance. Physiol. Genomics 2018, 50, 208–214. [Google Scholar] [CrossRef]
- Prabhakar, N.R.; Peng, Y.J. Oxygen Sensing by the Carotid Body: Past and Present. Adv. Exp. Med. Biol. 2017, 977, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.J.; Ortega, F.E.; Riegler, J.; Madison, D.V.; Krasnow, M.A. Oxygen regulation of breathing through an olfactory receptor activated by lactate. Nature 2015, 527, 240–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunham-Snary, K.J.; Wu, D.; Sykes, E.A.; Thakrar, A.; Parlow, L.R.G.; Mewburn, J.D.; Parlow, J.L.; Archer, S.L. Hypoxic Pulmonary Vasoconstriction: From Molecular Mechanisms to Medicine. Chest 2017, 151, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumb, A.B.; Slinger, P. Hypoxic pulmonary vasoconstriction: Physiology and anesthetic implications. Anesthesiology 2015, 122, 932–946. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef] [PubMed]
- Souma, T.; Nezu, M.; Nakano, D.; Yamazaki, S.; Hirano, I.; Sekine, H.; Dan, T.; Takeda, K.; Fong, G.H.; Nishiyama, A.; et al. Erythropoietin Synthesis in Renal Myofibroblasts Is Restored by Activation of Hypoxia Signaling. J. Am. Soc. Nephrol. 2016, 27, 428–438. [Google Scholar] [CrossRef]
- Suzuki, N.; Yamamoto, M. Roles of renal erythropoietin-producing (REP) cells in the maintenance of systemic oxygen homeostasis. Pflugers. Arch. 2016, 468, 3–12. [Google Scholar] [CrossRef]
- Yamawaki, T.; Ishikawa, H.; Mizuno, M.; Nakamura, H.; Shiro, Y.; Mizutani, Y. Regulatory Implications of Structural Changes in Tyr201 of the Oxygen Sensor Protein FixL. Biochemistry 2016, 55, 4027–4035. [Google Scholar] [CrossRef]
- Wright, G.S.A.; Saeki, A.; Hikima, T.; Nishizono, Y.; Hisano, T.; Kamaya, M.; Nukina, K.; Nishitani, H.; Nakamura, H.; Yamamoto, M.; et al. Architecture of the complete oxygen-sensing FixL-FixJ two-component signal transduction system. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Iwai, K.; Drake, S.K.; Wehr, N.B.; Weissman, A.M.; LaVaute, T.; Minato, N.; Klausner, R.D.; Levine, R.L.; Rouault, T.A. Iron-dependent oxidation, ubiquitination, and degradation of iron regulatory protein 2: Implications for degradation of oxidized proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 4924–4928. [Google Scholar] [CrossRef] [Green Version]
- Starkov, A.A.; Fiskum, G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Strielkov, I.; Pak, O.; Sommer, N.; Weissmann, N. Recent advances in oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction. J. Appl. Physiol. (1985) 2017, 123, 1647–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Saenz, P.; Lopez-Barneo, J. Physiology of the Carotid Body: From Molecules to Disease. Annu. Rev. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.H.; Semenza, G.L.; Bauer, C.; Marti, H.H. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am. J. Physiol. 1996, 271, C1172–C1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Torrelo, H.; Ortega-Saenz, P.; Macias, D.; Omura, M.; Zhou, T.; Matsunami, H.; Johnson, R.S.; Mombaerts, P.; Lopez-Barneo, J. The role of Olfr78 in the breathing circuit of mice. Nature 2018, 561, E33–E40. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H.; Callaway, E. Biologists who decoded how cells sense oxygen win medicine Nobel. Nature 2019, 574, 161–162. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Semenza, G. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J. Biol. Chem. 1993, 268, 21513–21518. [Google Scholar]
- Wang, G.; Jiang, B.; Rue, E.; Semenza, G. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Semenza, G. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Serendipity, Generosity, and Inspiration. Cell 2016, 167, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Percy, M.J.; Furlow, P.W.; Lucas, G.S.; Li, X.; Lappin, T.R.; McMullin, M.F.; Lee, F.S. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N. Engl. J. Med. 2008, 358, 162–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangaku, M.; Eckardt, K.U. Hypoxia and the HIF system in kidney disease. J. Mol. Med. (Berl) 2007, 85, 1325–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1 and human disease: One highly involved factor. Genes Dev. 2000, 14, 1983–1991. [Google Scholar] [PubMed]
- Semenza, G.L. HIF-1, O(2), and the 3 PHDs: How animal cells signal hypoxia to the nucleus. Cell 2001, 107, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Hirota, K.; Semenza, G.L. Regulation of angiogenesis by hypoxia-inducible factor 1. Crit. Rev. Oncol. Hematol. 2006, 59, 15–26. [Google Scholar] [CrossRef]
- Hirota, K. Involvement of hypoxia-inducible factors in the dysregulation of oxygen homeostasis in sepsis. Cardiovasc. Hematol. Disord. Drug Targets 2015, 15, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Hirota, K. Hypoxia-inducible factor 1, a master transcription factor of cellular hypoxic gene expression. J. Anesth. 2002, 16, 150–159. [Google Scholar] [CrossRef]
- Dang, C.V.; Semenza, G.L. Oncogenic alterations of metabolism. Trends Biochem Sci 1999, 24, 68–72. [Google Scholar] [CrossRef]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.E.; Simon, M.C. From stem cells to cancer stem cells: HIF takes the stage. Curr. Opin. Cell Biol. 2012, 24, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Hirano, I.; Pan, X.; Minegishi, N.; Yamamoto, M. Erythropoietin production in neuroepithelial and neural crest cells during primitive erythropoiesis. Nat. Commun. 2013, 4, 2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souma, T.; Suzuki, N.; Yamamoto, M. Renal erythropoietin-producing cells in health and disease. Front. Physiol. 2015, 6, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2alpha Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef]
- Wehn, P.M.; Rizzi, J.P.; Dixon, D.D.; Grina, J.A.; Schlachter, S.T.; Wang, B.; Xu, R.; Yang, H.; Du, X.; Han, G.; et al. Design and Activity of Specific Hypoxia-Inducible Factor-2alpha (HIF-2alpha) Inhibitors for the Treatment of Clear Cell Renal Cell Carcinoma: Discovery of Clinical Candidate ( S)-3-((2,2-Difluoro-1-hydroxy-7-(methylsulfonyl)-2,3-dihydro-1 H-inden-4-yl)oxy)-5-fluorobenzonitrile (PT2385). J. Med. Chem. 2018, 61, 9691–9721. [Google Scholar] [CrossRef]
- Srinivas, V.; Zhu, X.; Salceda, S.; Nakamura, R.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) is a non-heme iron protein. Implications for oxygen sensing. J. Biol. Chem. 1998, 273, 18019–18022. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, V.; Zhu, X.; Salceda, S.; Nakamura, R.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) is a non-heme iron protein. Implications for oxygen sensing. J. Biol. Chem. 1999, 274, 1180. [Google Scholar]
- Taylor, B.L.; Zhulin, I.B. PAS domains: Internal sensors of oxygen, redox potential, and light. Microbiol. Mol. Biol. Rev. 1999, 63, 479–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, J.B. Physiological Effects of Chronic Hypoxia. N. Engl. J. Med. 2017, 376, 1965–1971. [Google Scholar] [CrossRef] [PubMed]
- Fandrey, J.; Schodel, J.; Eckardt, K.U.; Katschinski, D.M.; Wenger, R.H. Now a Nobel gas: Oxygen. Pflugers. Arch. 2019. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Kallio, P.J.; Okamoto, K.; O’Brien, S.; Carrero, P.; Makino, Y.; Tanaka, H.; Poellinger, L. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998, 17, 6573–6586. [Google Scholar] [CrossRef]
- Luo, J.C.; Shibuya, M. A variant of nuclear localization signal of bipartite-type is required for the nuclear translocation of hypoxia inducible factors (1alpha, 2alpha and 3alpha). Oncogene 2001, 20, 1435–1444. [Google Scholar] [CrossRef] [Green Version]
- Depping, R.; Steinhoff, A.; Schindler, S.G.; Friedrich, B.; Fagerlund, R.; Metzen, E.; Hartmann, E.; Kohler, M. Nuclear translocation of hypoxia-inducible factors (HIFs): Involvement of the classical importin alpha/beta pathway. Biochim. Biophys. Acta. 2008, 1783, 394–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chachami, G.; Paraskeva, E.; Mingot, J.M.; Braliou, G.G.; Gorlich, D.; Simos, G. Transport of hypoxia-inducible factor HIF-1alpha into the nucleus involves importins 4 and 7. Biochem. Biophys. Res. Commun. 2009, 390, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.; Gleadle, J.; McNeill, L.; Hewitson, K.; O’Rourke, J.; Mole, D.; Mukherji, M.; Metzen, E.; Wilson, M.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Hirota, K.; Semenza, G.L. Regulation of hypoxia-inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochem Biophys. Res. Commun. 2005, 338, 610–616. [Google Scholar] [CrossRef]
- Hirsila, M.; Koivunen, P.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003, 278, 30772–30780. [Google Scholar] [CrossRef] [Green Version]
- Koivunen, P.; Hirsila, M.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J. Biol. Chem. 2004, 279, 9899–9904. [Google Scholar] [CrossRef] [Green Version]
- Metzen, E.; Wolff, M.; Fandrey, J.; Jelkmann, W. Pericellular PO2 and O2 consumption in monolayer cell cultures. Respir. Physiol. 1995, 100, 101–106. [Google Scholar] [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005, 1, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaelin, W. ROS: Really involved in oxygen sensing. Cell Metab. 2005, 1, 357–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, D.E.; Berra, E.; Gothie, E.; Roux, D.; Pouyssegur, J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J. Biol. Chem. 1999, 274, 32631–32637. [Google Scholar] [CrossRef] [Green Version]
- Conrad, P.W.; Freeman, T.L.; Beitner-Johnson, D.; Millhorn, D.E. EPAS1 trans-activation during hypoxia requires p42/p44 MAPK. J. Biol. Chem. 1999, 274, 33709–33713. [Google Scholar] [CrossRef] [Green Version]
- Kalousi, A.; Mylonis, I.; Politou, A.S.; Chachami, G.; Paraskeva, E.; Simos, G. Casein kinase 1 regulates human hypoxia-inducible factor HIF-1. J. Cell Sci. 2010, 123, 2976–2986. [Google Scholar] [CrossRef] [Green Version]
- Bencokova, Z.; Kaufmann, M.R.; Pires, I.M.; Lecane, P.S.; Giaccia, A.J.; Hammond, E.M. ATM activation and signaling under hypoxic conditions. Mol. Cell Biol. 2009, 29, 526–537. [Google Scholar] [CrossRef] [Green Version]
- Flugel, D.; Gorlach, A.; Michiels, C.; Kietzmann, T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol. Cell Biol. 2007, 27, 3253–3265. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Yao, Y.; Lu, L.; Costa, M.; Dai, W. Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF-1alpha. J. Biol. Chem. 2010, 285, 38944–38950. [Google Scholar] [CrossRef] [Green Version]
- Bullen, J.W.; Tchernyshyov, I.; Holewinski, R.J.; DeVine, L.; Wu, F.; Venkatraman, V.; Kass, D.L.; Cole, R.N.; Van Eyk, J.; Semenza, G.L. Protein kinase A-dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci. Signal. 2016, 9, ra56. [Google Scholar] [CrossRef] [Green Version]
- McNamee, E.N.; Vohwinkel, C.; Eltzschig, H.K. Hydroxylation-independent HIF-1alpha stabilization through PKA: A new paradigm for hypoxia signaling. Sci. Signal. 2016, 9, fs11. [Google Scholar] [CrossRef] [Green Version]
- Gkotinakou, I.M.; Befani, C.; Simos, G.; Liakos, P. ERK1/2 phosphorylates HIF-2alpha and regulates its activity by controlling its CRM1-dependent nuclear shuttling. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Pangou, E.; Befani, C.; Mylonis, I.; Samiotaki, M.; Panayotou, G.; Simos, G.; Liakos, P. HIF-2alpha phosphorylation by CK1delta promotes erythropoietin secretion in liver cancer cells under hypoxia. J. Cell Sci. 2016, 129, 4213–4226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dioum, E.M.; Chen, R.; Alexander, M.S.; Zhang, Q.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 2009, 324, 1289–1293. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.G.; Kong, G.; Lee, M.O. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1alpha protein by recruiting histone deacetylase 1. EMBO J. 2006, 25, 1231–1241. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xu, Y.; Jiao, H.; Wang, W.; Mei, Z.; Chen, G. Sumoylation of hypoxia inducible factor-1alpha and its significance in cancer. Sci. China Life Sci. 2014, 57, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.H.; Jeong, J.W.; Park, J.A.; Kim, S.H.; Bae, M.K.; Choi, S.J.; Kim, K.W. Sumoylation increases HIF-1alpha stability and its transcriptional activity. Biochem. Biophys. Res. Commun. 2004, 324, 394–400. [Google Scholar] [CrossRef]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.; Li, C.Y. Regulation of HIF-1alpha stability through S-nitrosylation. Mol. Cell 2007, 26, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, J.H.; Li, S.H.; Park, H.S.; Park, J.W.; Lee, B.; Chun, Y.S. Hypoxia-inducible factor alpha subunit stabilization by NEDD8 conjugation is reactive oxygen species-dependent. J. Biol. Chem. 2011, 286, 6963–6970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ema, M.; Hirota, K.; Mimura, J.; Abe, H.; Yodoi, J.; Sogawa, K.; Poellinger, L.; Fujii-Kuriyama, Y. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: Their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999, 18, 1905–1914. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Schumacker, P.T. Hypoxia, anoxia, and O2 sensing: The search continues. Am. J. Physiol. Lung Cell Mol. Physiol 2002, 283, L918–L921. [Google Scholar] [CrossRef] [Green Version]
- Yuan, G.; Khan, S.A.; Luo, W.; Nanduri, J.; Semenza, G.L.; Prabhakar, N.R. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J. Cell Physiol. 2011, 226, 2925–2933. [Google Scholar] [CrossRef] [Green Version]
- Nanduri, J.; Makarenko, V.; Reddy, V.D.; Yuan, G.; Pawar, A.; Wang, N.; Khan, S.A.; Zhang, X.; Kinsman, B.; Peng, Y.J.; et al. Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 2515–2520. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, N.R.; Peng, Y.J.; Yuan, G.; Nanduri, J. Reactive oxygen radicals and gaseous transmitters in carotid body activation by intermittent hypoxia. Cell Tissue Res. 2018, 372, 427–431. [Google Scholar] [CrossRef]
- Yuan, G.; Peng, Y.J.; Khan, S.A.; Nanduri, J.; Singh, A.; Vasavda, C.; Semenza, G.L.; Kumar, G.K.; Snyder, S.H.; Prabhakar, N.R. H2S production by reactive oxygen species in the carotid body triggers hypertension in a rodent model of sleep apnea. Sci. Signal. 2016, 9, ra80. [Google Scholar] [CrossRef] [Green Version]
- Nanduri, J.; Peng, Y.J.; Wang, N.; Khan, S.A.; Semenza, G.L.; Kumar, G.K.; Prabhakar, N.R. Epigenetic regulation of redox state mediates persistent cardiorespiratory abnormalities after long-term intermittent hypoxia. J. Physiol. 2017, 595, 63–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakar, N.R.; Semenza, G.L. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol. Rev. 2012, 92, 967–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000, 14, 34–44. [Google Scholar] [PubMed]
- Semenza, G.L. VHL and p53: Tumor suppressors team up to prevent cancer. Mol. Cell 2006, 22, 437–439. [Google Scholar] [CrossRef]
- An, W.G.; Kanekal, M.; Simon, M.C.; Maltepe, E.; Blagosklonny, M.V.; Neckers, L.M. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature 1998, 392, 405–408. [Google Scholar] [CrossRef]
- Amelio, I.; Mancini, M.; Petrova, V.; Cairns, R.A.; Vikhreva, P.; Nicolai, S.; Marini, A.; Antonov, A.A.; Le Quesne, J.; Baena Acevedo, J.D.; et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E10869–E10878. [Google Scholar] [CrossRef] [Green Version]
- D’Orazi, G.; Cirone, M. Mutant p53 and Cellular Stress Pathways: A Criminal Alliance That Promotes Cancer Progression. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, R.; Hirota, K.; Fan, F.; Jung, Y.D.; Ellis, L.M.; Semenza, G.L. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J. Biol. Chem. 2002, 277, 38205–38211. [Google Scholar] [CrossRef] [Green Version]
- Treins, C.; Giorgetti-Peraldi, S.; Murdaca, J.; Semenza, G.L.; Van Obberghen, E. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J. Biol. Chem. 2002, 277, 27975–27981. [Google Scholar] [CrossRef] [Green Version]
- Hirota, K.; Fukuda, R.; Takabuchi, S.; Kizaka-Kondoh, S.; Adachi, T.; Fukuda, K.; Semenza, G.L. Induction of hypoxia-inducible factor 1 activity by muscarinic acetylcholine receptor signaling. J. Biol. Chem. 2004, 279, 41521–41528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Kelly, B.; Semenza, G.L. Vascular endothelial growth factor gene expression in colon cancer cells exposed to prostaglandin E2 is mediated by hypoxia-inducible factor 1. Cancer Res. 2003, 63, 2330–2334. [Google Scholar] [PubMed]
- Suzuki, K.; Nishi, K.; Takabuchi, S.; Kai, S.; Matsuyama, T.; Kurosawa, S.; Adachi, T.; Maruyama, T.; Fukuda, K.; Hirota, K. Differential roles of prostaglandin E-type receptors in activation of hypoxia-inducible factor 1 by prostaglandin E1 in vascular-derived cells under non-hypoxic conditions. PeerJ 2013, 1, e220. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Jiang, B.H.; Liu, L.Z. PI3K/PTEN signaling in tumorigenesis and angiogenesis. Biochim. Biophys. Acta 2008, 1784, 150–158. [Google Scholar] [CrossRef]
- Daijo, H.; Hoshino, Y.; Kai, S.; Suzuki, K.; Nishi, K.; Matsuo, Y.; Harada, H.; Hirota, K. Cigarette smoke reversibly activates hypoxia-inducible factor 1 in a reactive oxygen species-dependent manner. Sci. Rep. 2016, 6, 34424. [Google Scholar] [CrossRef] [Green Version]
- Peyssonnaux, C.; Datta, V.; Cramer, T.; Doedens, A.; Theodorakis, E.A.; Gallo, R.L.; Hurtado-Ziola, N.; Nizet, V.; Johnson, R.S. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J. Clin. Invest. 2005, 115, 1806–1815. [Google Scholar] [CrossRef] [Green Version]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef]
- Cramer, T.; Johnson, R.S. A novel role for the hypoxia inducible transcription factor HIF-1alpha: Critical regulation of inflammatory cell function. Cell Cycle 2003, 2, 192–193. [Google Scholar] [CrossRef] [Green Version]
- Nishi, K.; Oda, T.; Takabuchi, S.; Oda, S.; Fukuda, K.; Adachi, T.; Semenza, G.L.; Shingu, K.; Hirota, K. LPS induces hypoxia-inducible factor 1 activation in macrophage-differentiated cells in a reactive oxygen species-dependent manner. Antioxid Redox Signal. 2008, 10, 983–996. [Google Scholar] [CrossRef]
- Wang, F.; Sekine, H.; Kikuchi, Y.; Takasaki, C.; Miura, C.; Heiwa, O.; Shuin, T.; Fujii-Kuriyama, Y.; Sogawa, K. HIF-1alpha-prolyl hydroxylase: Molecular target of nitric oxide in the hypoxic signal transduction pathway. Biochem. Biophys. Res. Commun. 2002, 295, 657–662. [Google Scholar] [CrossRef]
- Sandau, K.B.; Fandrey, J.; Brune, B. Accumulation of HIF-1alpha under the influence of nitric oxide. Blood 2001, 97, 1009–1015. [Google Scholar] [CrossRef]
- Sandau, K.B.; Faus, H.G.; Brune, B. Induction of hypoxia-inducible-factor 1 by nitric oxide is mediated via the PI 3K pathway. Biochem. Biophys. Res. Commun. 2000, 278, 263–267. [Google Scholar] [CrossRef]
- Metzen, E.; Zhou, J.; Jelkmann, W.; Fandrey, J.; Brune, B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol. Biol. Cell 2003, 14, 3470–3481. [Google Scholar] [CrossRef] [Green Version]
- Brune, B.; Zhou, J. The role of nitric oxide (NO) in stability regulation of hypoxia inducible factor-1alpha (HIF-1alpha). Curr. Med. Chem. 2003, 10, 845–855. [Google Scholar] [CrossRef]
- Kasuno, K.; Takabuchi, S.; Fukuda, K.; Kizaka-Kondoh, S.; Yodoi, J.; Adachi, T.; Semenza, G.L.; Hirota, K. Nitric oxide induces hypoxia-inducible factor 1 activation that is dependent on MAPK and phosphatidylinositol 3-kinase signaling. J. Biol. Chem. 2004, 279, 2550–2558. [Google Scholar] [CrossRef] [Green Version]
- Palmer, L.A.; Gaston, B.; Johns, R.A. Normoxic stabilization of hypoxia-inducible factor-1 expression and activity: Redox-dependent effect of nitrogen oxides. Mol. Pharmacol. 2000, 58, 1197–1203. [Google Scholar] [CrossRef]
- Mateo, J.; Garcia-Lecea, M.; Cadenas, S.; Hernandez, C.; Moncada, S. Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem. J. 2003, 376, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Sumbayev, V.V.; Yasinska, I.M. Peroxynitrite as an alternative donor of oxygen in HIF-1alpha proline hydroxylation under low oxygen availability. Free Radic. Res. 2006, 40, 631–635. [Google Scholar] [CrossRef]
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159. [Google Scholar] [CrossRef] [Green Version]
- Dehne, N.; Brune, B. HIF-1 in the inflammatory microenvironment. Exp. Cell Res. 2009, 315, 1791–1797. [Google Scholar] [CrossRef] [PubMed]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Colgan, S.P.; Taylor, C.T. Hypoxia: An alarm signal during intestinal inflammation. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 281–287. [Google Scholar] [CrossRef] [Green Version]
- Walmsley, S.R.; Cadwallader, K.A.; Chilvers, E.R. The role of HIF-1alpha in myeloid cell inflammation. Trends Immunol. 2005, 26, 434–439. [Google Scholar] [CrossRef]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Tanaka, T.; Yamamoto, T.; Noiri, E.; Miyata, T.; Inagi, R.; Fujita, T.; Nangaku, M. Hypoperfusion of peritubular capillaries induces chronic hypoxia before progression of tubulointerstitial injury in a progressive model of rat glomerulonephritis. J. Am. Soc. Nephrol. 2004, 15, 1574–1581. [Google Scholar] [CrossRef] [Green Version]
- Manotham, K.; Tanaka, T.; Matsumoto, M.; Ohse, T.; Miyata, T.; Inagi, R.; Kurokawa, K.; Fujita, T.; Nangaku, M. Evidence of tubular hypoxia in the early phase in the remnant kidney model. J. Am. Soc. Nephrol. 2004, 15, 1277–1288. [Google Scholar] [CrossRef]
- Tanaka, T.; Miyata, T.; Inagi, R.; Fujita, T.; Nangaku, M. Hypoxia in renal disease with proteinuria and/or glomerular hypertension. Am. J. Pathol. 2004, 165, 1979–1992. [Google Scholar] [CrossRef] [Green Version]
- Katavetin, P.; Miyata, T.; Inagi, R.; Tanaka, T.; Sassa, R.; Ingelfinger, J.R.; Fujita, T.; Nangaku, M. High glucose blunts vascular endothelial growth factor response to hypoxia via the oxidative stress-regulated hypoxia-inducible factor/hypoxia-responsible element pathway. J. Am. Soc. Nephrol. 2006, 17, 1405–1413. [Google Scholar] [CrossRef] [Green Version]
- Katavetin, P.; Inagi, R.; Miyata, T.; Tanaka, T.; Sassa, R.; Ingelfinger, J.R.; Fujita, T.; Nangaku, M. Albumin suppresses vascular endothelial growth factor via alteration of hypoxia-inducible factor/hypoxia-responsive element pathway. Biochem. Biophys. Res. Commun. 2008, 367, 305–310. [Google Scholar] [CrossRef]
- Izuhara, Y.; Nangaku, M.; Inagi, R.; Tominaga, N.; Aizawa, T.; Kurokawa, K.; van Ypersele de Strihou, C.; Miyata, T. Renoprotective properties of angiotensin receptor blockers beyond blood pressure lowering. J. Am. Soc. Nephrol. 2005, 16, 3631–3641. [Google Scholar] [CrossRef] [Green Version]
- Hirota, K. An intimate crosstalk between iron homeostasis and oxygen metabolism regulated by the hypoxia-inducible factors (HIFs). Free Radic. Biol. Med. 2019, 133, 118–129. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J. Clin. Invest. 2007, 117, 1926–1932. [Google Scholar] [CrossRef] [Green Version]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Simpson, R.J.; McKie, A.T. Regulation of intestinal iron absorption: The mucosa takes control? Cell Metab. 2009, 10, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Ganz, T. Regulation of iron metabolism by hepcidin. Annu Rev. Nutr. 2006, 26, 323–342. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Hao, C.; Liu, B.C.; Lin, H.; Wang, C.; Xing, C.; Liang, X.; Jiang, G.; Liu, Z.; Li, X.; et al. Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis. N. Engl. J. Med. 2019, 381, 1011–1022. [Google Scholar] [CrossRef]
- Kaplan, J. Roxadustat and Anemia of Chronic Kidney Disease. N. Engl. J. Med. 2019, 381, 1070–1072. [Google Scholar] [CrossRef]
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N. Engl. J. Med. 2019, 381, 1001–1010. [Google Scholar] [CrossRef]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattigan, Y.I.; Patel, B.B.; Ackerstaff, E.; Sukenick, G.; Koutcher, J.A.; Glod, J.W.; Banerjee, D. Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp. Cell Res. 2012, 318, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Haase, V.H. Oxygen regulates epithelial-to-mesenchymal transition: Insights into molecular mechanisms and relevance to disease. Kidney Int. 2009, 76, 492–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.J.; Semenza, G.L.; Zhang, H.F. Hypoxia-inducible factor 1 and breast cancer metastasis. J. Zhejiang Univ. Sci. B 2015, 16, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Molecular mechanisms mediating metastasis of hypoxic breast cancer cells. Trends Mol. Med. 2012, 18, 534–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suetrong, B.; Walley, K.R. Lactic Acidosis in Sepsis: It’s Not All Anaerobic: Implications for Diagnosis and Management. Chest 2016, 149, 252–261. [Google Scholar] [CrossRef]
- Cray, S.H.; Robinson, B.H.; Cox, P.N. Lactic acidemia and bradyarrhythmia in a child sedated with propofol. Crit. Care Med. 1998, 26, 2087–2092. [Google Scholar] [CrossRef]
- Finsterer, J.; Frank, M. Propofol Is Mitochondrion-Toxic and May Unmask a Mitochondrial Disorder. J. Child. Neurol. 2016, 31, 1489–1494. [Google Scholar] [CrossRef]
- Krajcova, A.; Waldauf, P.; Andel, M.; Duska, F. Propofol infusion syndrome: A structured review of experimental studies and 153 published case reports. Crit. Care 2015, 19, 398. [Google Scholar] [CrossRef] [Green Version]
- Sumi, C.; Okamoto, A.; Tanaka, H.; Kusunoki, M.; Shoji, T.; Uba, T.; Adachi, T.; Iwai, T.; Nishi, K.; Harada, H.; et al. Suppression of mitochondrial oxygen metabolism mediated by the transcription factor HIF-1 alleviates propofol-induced cell toxicity. Sci. Rep. 2018, 8, 8987. [Google Scholar] [CrossRef]
- Abe, H.; Semba, H.; Takeda, N. The Roles of Hypoxia Signaling in the Pathogenesis of Cardiovascular Diseases. J. Atheroscler. Thromb. 2017, 24, 884–894. [Google Scholar] [CrossRef] [Green Version]
- Abe, H.; Takeda, N.; Isagawa, T.; Semba, H.; Nishimura, S.; Morioka, M.S.; Nakagama, Y.; Sato, T.; Soma, K.; Koyama, K.; et al. Macrophage hypoxia signaling regulates cardiac fibrosis via Oncostatin M. Nat. Commun. 2019, 10, 2824. [Google Scholar] [CrossRef]
- Semba, H.; Takeda, N.; Isagawa, T.; Sugiura, Y.; Honda, K.; Wake, M.; Miyazawa, H.; Yamaguchi, Y.; Miura, M.; Jenkins, D.M.; et al. HIF-1alpha-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat. Commun. 2016, 7, 11635. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Minamino, T.; Toko, H.; Miyauchi, H.; Orimo, M.; Qin, Y.; Akazawa, H.; Tateno, K.; Kayama, Y.; Harada, M.; et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007, 446, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Mirtschink, P.; Krishnan, J.; Grimm, F.; Sarre, A.; Horl, M.; Kayikci, M.; Fankhauser, N.; Christinat, Y.; Cortijo, C.; Feehan, O.; et al. HIF-driven SF3B1 induces KHK-C to enforce fructolysis and heart disease. Nature 2015, 522, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Caniggia, I.; Winter, J.; Lye, S.J.; Post, M. Oxygen and placental development during the first trimester: Implications for the pathophysiology of pre-eclampsia. Placenta 2000, 21, S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Red-Horse, K.; Zhou, Y.; Genbacev, O.; Prakobphol, A.; Foulk, R.; McMaster, M.; Fisher, S.J. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J. Clin. Invest. 2004, 114, 744–754. [Google Scholar] [CrossRef]
- Cross, J.C.; Baczyk, D.; Dobric, N.; Hemberger, M.; Hughes, M.; Simmons, D.G.; Yamamoto, H.; Kingdom, J.C. Genes, development and evolution of the placenta. Placenta 2003, 24, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Caniggia, I.; Mostachfi, H.; Winter, J.; Gassmann, M.; Lye, S.J.; Kuliszewski, M.; Post, M. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3). J. Clin. Invest. 2000, 105, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Hirota, Y. Progesterone governs endometrial proliferation-differentiation switching and blastocyst implantation. Endocr. J. 2019, 66, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Fukui, Y.; Hirota, Y.; Matsuo, M.; Gebril, M.; Akaeda, S.; Hiraoka, T.; Osuga, Y. Uterine receptivity, embryo attachment, and embryo invasion: Multistep processes in embryo implantation. Reprod. Med. Biol. 2019, 18, 234–240. [Google Scholar] [CrossRef]
- Matsumoto, L.; Hirota, Y.; Saito-Fujita, T.; Takeda, N.; Tanaka, T.; Hiraoka, T.; Akaeda, S.; Fujita, H.; Shimizu-Hirota, R.; Igaue, S.; et al. HIF2alpha in the uterine stroma permits embryo invasion and luminal epithelium detachment. J. Clin. Invest. 2018, 128, 3186–3197. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Makino, Y.; Okamoto, K.; Poellinger, L.; Ohnuma, K.; Morimoto, C.; Tanaka, H. TCR engagement increases hypoxia-inducible factor-1 alpha protein synthesis via rapamycin-sensitive pathway under hypoxic conditions in human peripheral T cells. J. Immunol. 2005, 174, 7592–7599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makino, Y.; Nakamura, H.; Ikeda, E.; Ohnuma, K.; Yamauchi, K.; Yabe, Y.; Poellinger, L.; Okada, Y.; Morimoto, C.; Tanaka, H. Hypoxia-inducible factor regulates survival of antigen receptor-driven T cells. J. Immunol. 2003, 171, 6534–6540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, T.; Hirota, K.; Nishi, K.; Takabuchi, S.; Oda, S.; Yamada, H.; Arai, T.; Fukuda, K.; Kita, T.; Adachi, T.; et al. Activation of hypoxia-inducible factor 1 during macrophage differentiation. Am. J. Physiol. Cell Physiol. 2006, 291, C104–C113. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, T.; Han, J.H.; Devkota, S.; Lee, H.W. Emerging Paradigm of Crosstalk between Autophagy and the Ubiquitin-Proteasome System. Mol. Cells 2017, 40, 897–905. [Google Scholar] [CrossRef]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Chourasia, A.H.; Macleod, K.F. Tumor suppressor functions of BNIP3 and mitophagy. Autophagy 2015, 11, 1937–1938. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Xiong, X.; Jia, L.; Sun, Y. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis. 2012, 3, e386. [Google Scholar] [CrossRef]
- Wolff, N.C.; Vega-Rubin-de-Celis, S.; Xie, X.J.; Castrillon, D.H.; Kabbani, W.; Brugarolas, J. Cell-type-dependent regulation of mTORC1 by REDD1 and the tumor suppressors TSC1/TSC2 and LKB1 in response to hypoxia. Mol. Cell Biol. 2011, 31, 1870–1884. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Km (µM) | ||||||
|---|---|---|---|---|---|---|---|
| Gene | Protein | Pro-402 | Pro-564 | O2 | α-ketoglutarate | Ascorbate | Fe(II) |
| EGLN1 | PHD2/HPH-2 | + | + | 230 | 60 | 170 | 0.03 |
| EGLN2 | PHD-1/HPH-3 | + | + | 250 | 60 | 180 | 0.1 |
| EGLN3 | PHD3/HPH-1 | − | + | 230 | 55 | 140 | 0.03 |
| HIF1AN | FIH-1 | Asp-803 | 90 | 25 | 260 | 0.5 | |
| P4HA1 | C-P4H-I | collagen | 40 | 20 | 300 | ||
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirota, K. Basic Biology of Hypoxic Responses Mediated by the Transcription Factor HIFs and Its Implication for Medicine. Biomedicines 2020, 8, 32. https://doi.org/10.3390/biomedicines8020032
Hirota K. Basic Biology of Hypoxic Responses Mediated by the Transcription Factor HIFs and Its Implication for Medicine. Biomedicines. 2020; 8(2):32. https://doi.org/10.3390/biomedicines8020032
Chicago/Turabian StyleHirota, Kiichi. 2020. "Basic Biology of Hypoxic Responses Mediated by the Transcription Factor HIFs and Its Implication for Medicine" Biomedicines 8, no. 2: 32. https://doi.org/10.3390/biomedicines8020032