Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer




Abstract
:
1. Introduction
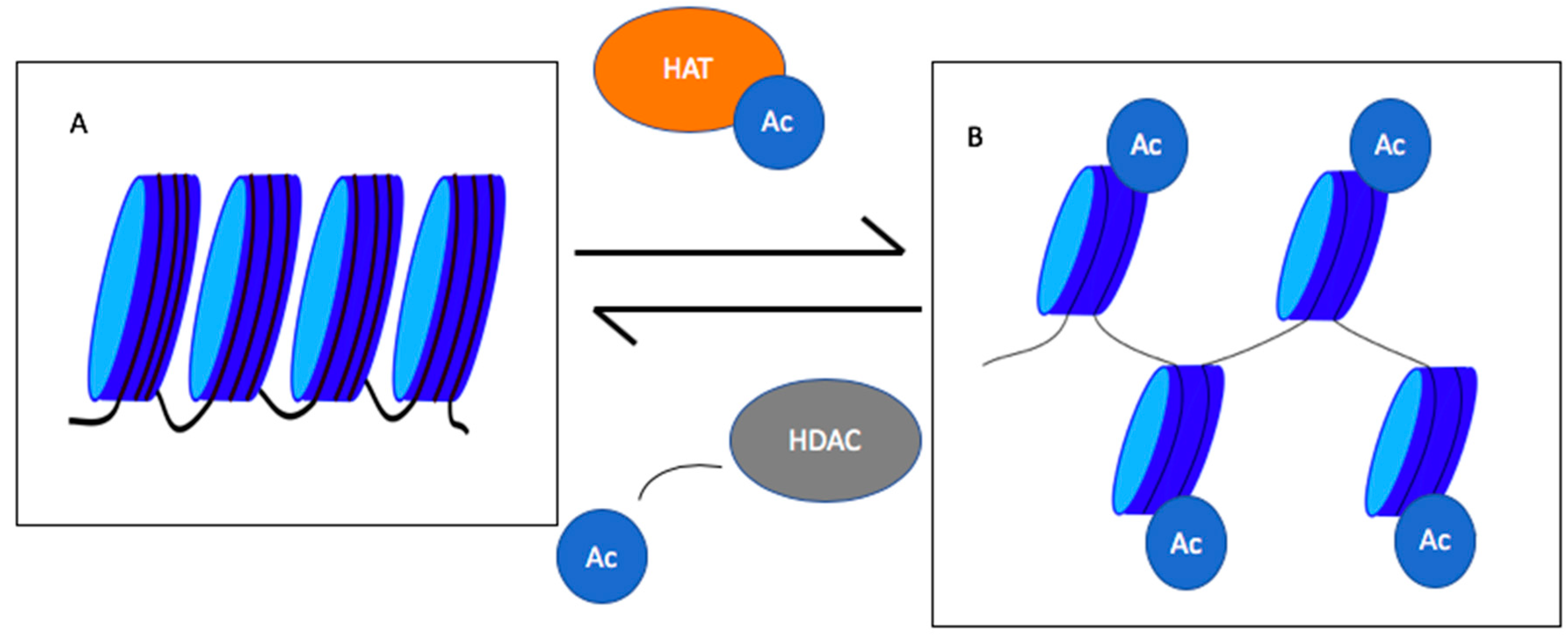
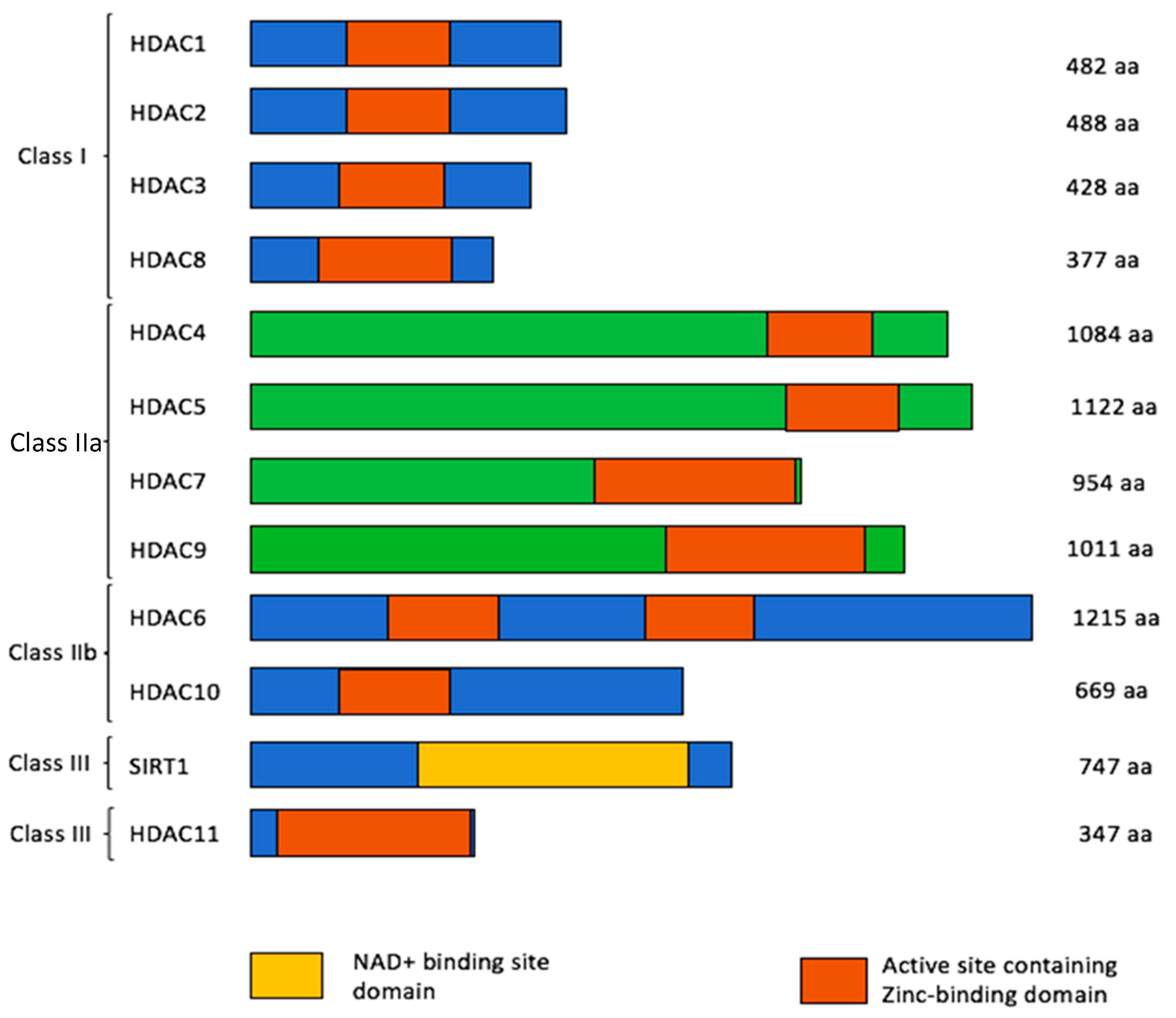
2. Histone Deacetylases (HDACs)
3. HDAC Inhibitors
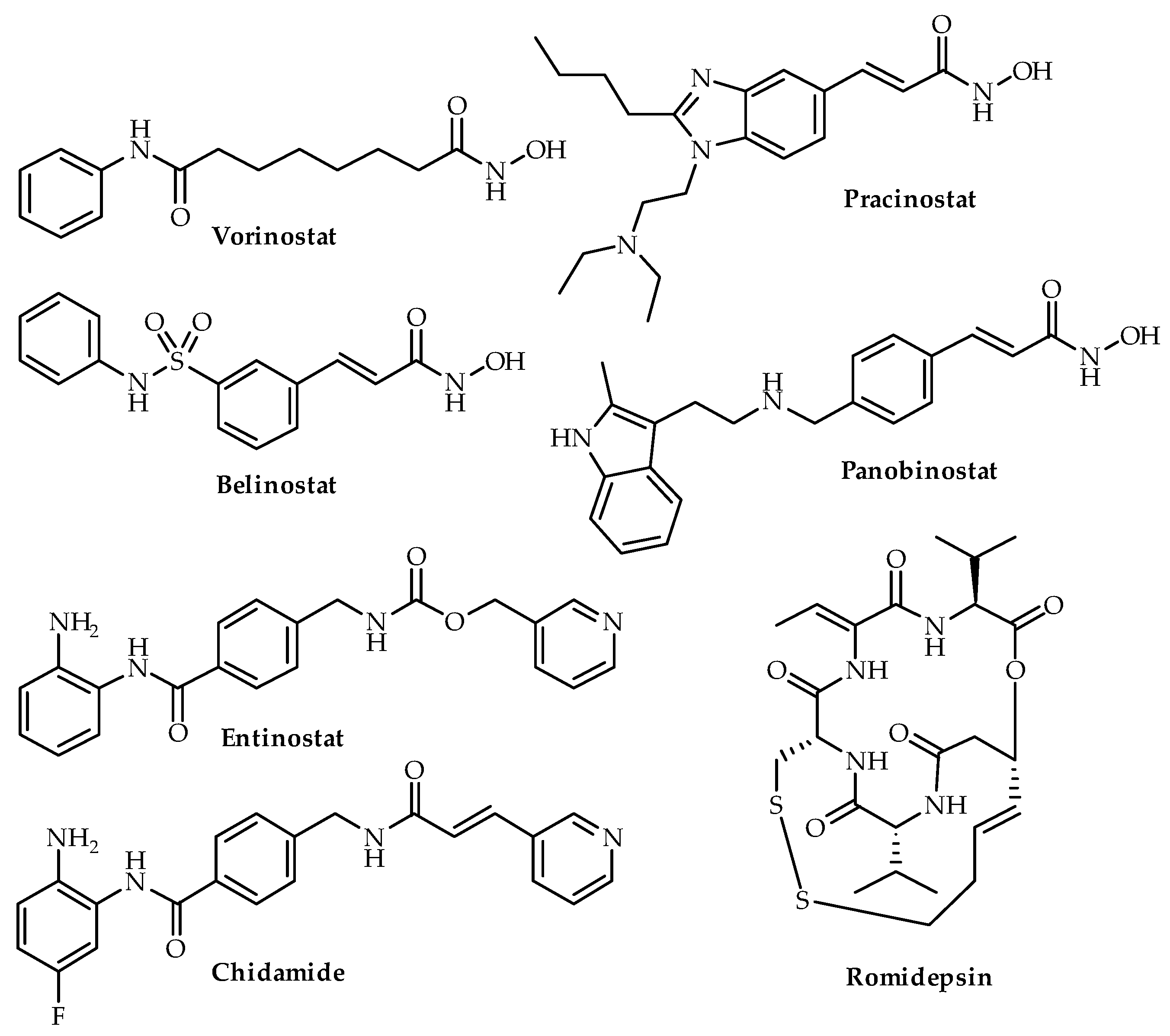
3.1. Vorinostat (SAHA)
3.2. Panobinostat
3.3. Romidepsin
3.4. Pracinostat
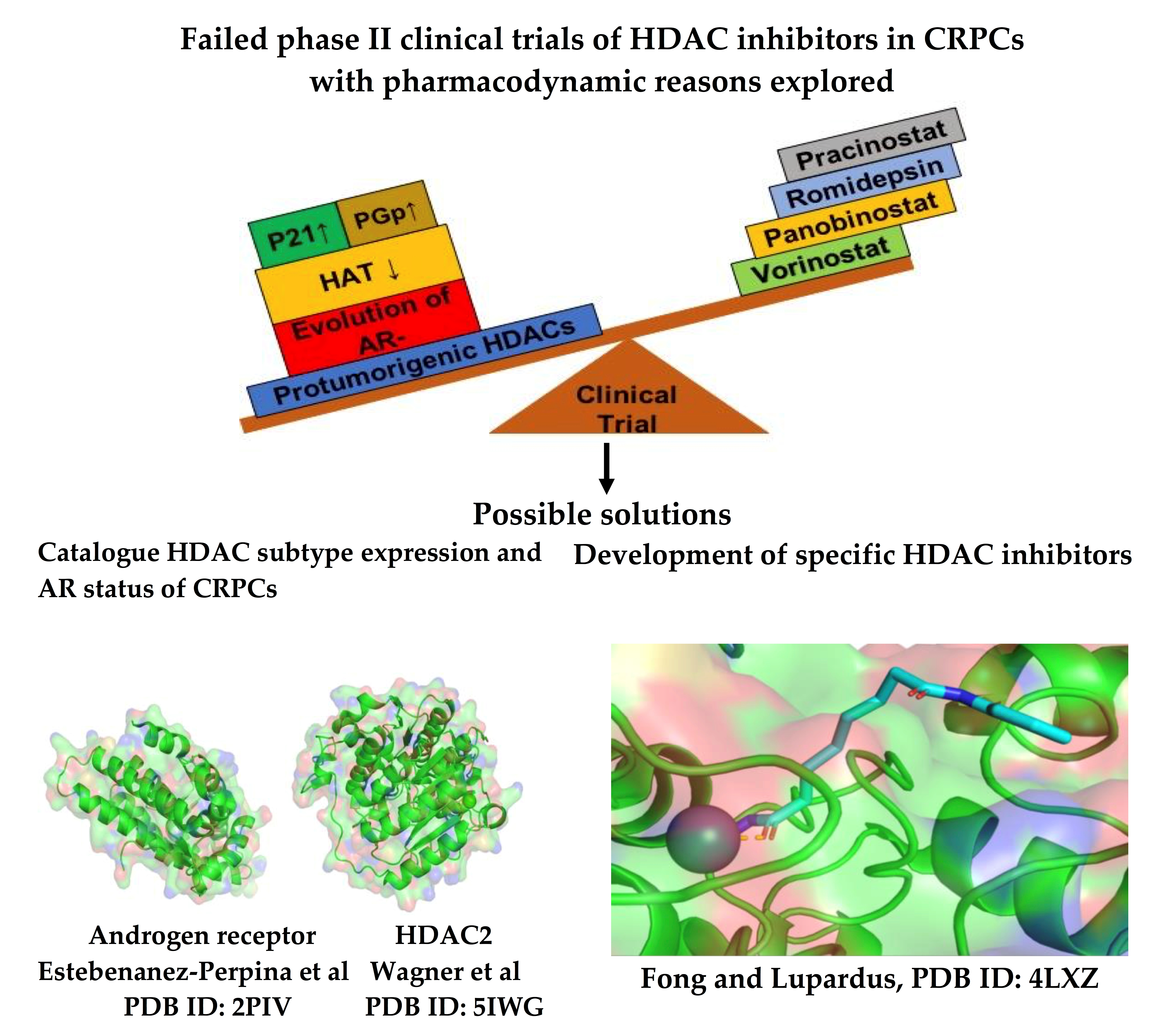
4. Pharmacodynamic Rationale for Treatment Failure Using HDAC Inhibitors
4.1. AR-Negative Prostate Cancers
4.2. Other Cellular Targets of HDACs
4.3. Resistance Mechanisms
4.4. P-Glycoprotein
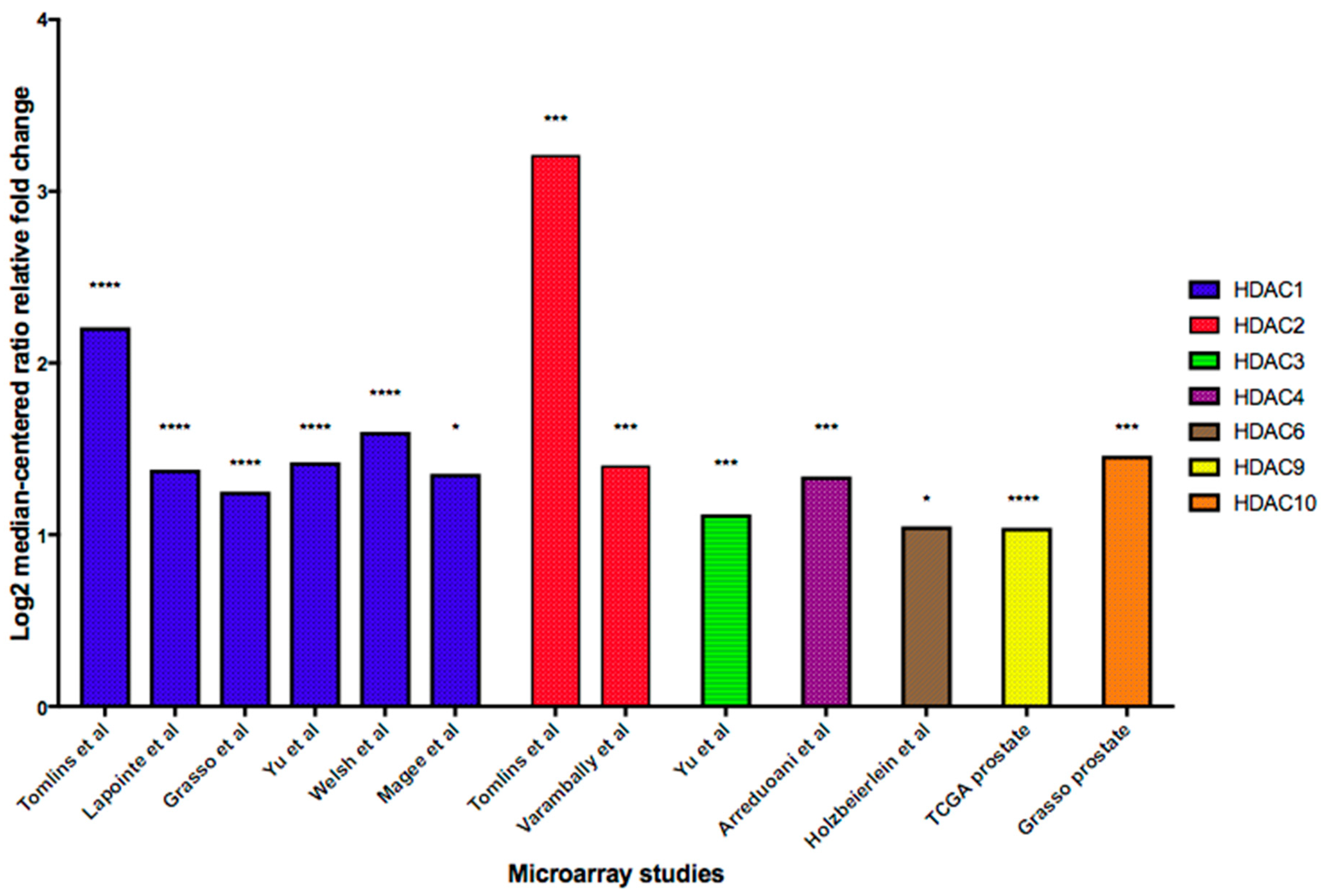
4.5. HDAC Upregulation
4.6. Histone Acetyltransferases (HATs) Downregulation
4.7. P21 Upregulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CRPC | Castration Resistant Prostate Cancer |
| HDAC | Histone Deacetylase |
| HAT | Histone Acetyl Transferase |
| DNA | Deoxyribonucleic Acid |
| NF- kB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| FoxO1 | Forkhead box protein O1 |
| PSA | Prostate Specific Antigen |
| HSP-90 | Heat Shock Protein-90 |
| SAHA | Suberoylanilide Hydroxamic acid |
| IL6 | Interleukin 6 |
| LBH589 | Panobinostat |
| LAQ824 | Dacinostat |
| FDA | Food and Drug Administration |
| AR | Androgen Receptor |
| RECIST | Response Evaluation Criteria in Solid Tumors |
| CTC | Circulating Tumor Cell |
| ERG | ETS-related Gene |
| PTEN | Phosphatase and Tensin homolog |
| NE | Neuroendocrine |
| FGF-8 | Fibroblast Growth Factor-8 |
| MAPK | Mitogen-Activated Protein Kinase |
| EMT | Epithelial-Mesenchymal Transition |
| ZEB1 | Zinc finger E-box-binding homeobox 1 |
| MMP2 | Matrix Metalloproteinase-2 |
| TSA | Trichostatin A |
| VPA | Valproic Acid |
| HIF-1α | Hypoxia-inducible Factor 1-alpha |
| VEGFR-2 | Vascular Endothelial Growth Factor Receptor 2 |
| PEDF | Pigment Epithelium-derived Factor |
| KLF-4 | Kruppel-like Factor 4 |
| ChIP | Chromatin immunoprecipitation |
| FGF2 | Fibroblast Growth Factor 2 |
| PDGF-B | Platelet-derived Growth Factor Beta |
| MEF2 | Myocyte Enhancer Factor-2 |
| CSC | Cancer Stem-like Cells |
| AMP | Adenosine Monophosphate |
| CREB | cAMP Response Element-binding Protein |
| PTPN22 | Protein Tyrosine Phosphatase, Non-receptor type 22 |
| MMR | Mismatch Repair |
| WNT | Wingless-related Integration Site |
| PPAR | Peroxisome Proliferator-activated Receptor |
| BMP-4 | Bone Morphogenetic Protein 4 |
| P-Gp | P-Glycoproteins |
| MDR1 | Multi-drug Resistance-1 |
| CsA | Cyclosporin A |
| TRAMP | Transgenic Adenocarcinoma of the Mouse Prostate |
| MD | Moderately Differentiated |
References
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, D.; Vashistha, V.; Isharwal, S.; Sediqe, S.A.; Lin, M.-F. Histone deacetylase inhibitors in castration-resistant prostate cancer: Molecular mechanism of action and recent clinical trials. Ther. Adv. Urol. 2015, 7, 388–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damodaran, S.; Kyriakopoulos, C.E.; Jarrard, D.F. Newly Diagnosed Metastatic Prostate Cancer: Has the Paradigm Changed? Urol. Clin. North Am. 2017, 44, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524. [Google Scholar] [PubMed]
- Saraon, P.; Drabovich, A.P.; Jarvi, K.A.; Diamandis, E.P. Mechanisms of Androgen-Independent Prostate Cancer. EJIFCC 2014, 25, 42–54. [Google Scholar]
- Sartor, A.O. Progression of metastatic castrate-resistant prostate cancer: Impact of therapeutic intervention in the post-docetaxel space. J. Hematol. Oncol. 2011, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Jiang, X.; Liang, X.; Jiang, G. Molecular and cellular mechanisms of castration resistant prostate cancer (Review). Oncol. Lett. 2018. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.N.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.-W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [Green Version]
- Drogaris, P.; Villeneuve, V.; Pomiès, C.; Lee, E.-H.; Bourdeau, V.; Bonneil, É.; Ferbeyre, G.; Verreault, A.; Thibault, P. Histone Deacetylase Inhibitors Globally Enhance H3/H4 Tail Acetylation Without Affecting H3 Lysine 56 Acetylation. Sci. Rep. 2012, 2, 220. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Lee, E.Y.H.P.; Muller, W.J. Oncogenes and Tumor Suppressor Genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a003236. [Google Scholar] [CrossRef] [Green Version]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-J.; Bae, S.-C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Vadivelan, S.; Sinha, B.N.; Rambabu, G.; Boppana, K.; Jagarlapudi, S.A.R.P. Pharmacophore modeling and virtual screening studies to design some potential histone deacetylase inhibitors as new leads. J. Mol. Graph. Model. 2008, 26, 935–946. [Google Scholar] [CrossRef]
- Federico, M.; Bagella, L. Histone Deacetylase Inhibitors in the Treatment of Hematological Malignancies and Solid Tumors. J. Biomed. Biotechnol. 2011, 2011, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009, 280, 168–176. [Google Scholar] [CrossRef]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia N. Y. N 2004, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.; Ahmad, A.; Bao, B.; Li, Y.; Banerjee, S.; Sarkar, F.H. Histone Deacetylase Inhibitors Induce Epithelial-to-Mesenchymal Transition in Prostate Cancer Cells. PLoS ONE 2012, 7, e45045. [Google Scholar] [CrossRef]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef]
- Zimberlin, C.D.; Lancini, C.; Sno, R.; Rosekrans, S.L.; McLean, C.M.; Vlaming, H.; van den Brink, G.R.; Bots, M.; Medema, J.P.; Dannenberg, J.-H. HDAC1 and HDAC2 collectively regulate intestinal stem cell homeostasis. FASEB J. 2015, 29, 2070–2080. [Google Scholar] [CrossRef]
- Jamaladdin, S.; Kelly, R.D.W.; O’Regan, L.; Dovey, O.M.; Hodson, G.E.; Millard, C.J.; Portolano, N.; Fry, A.M.; Schwabe, J.W.R.; Cowley, S.M. Histone deacetylase (HDAC) 1 and 2 are essential for accurate cell division and the pluripotency of embryonic stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 9840–9845. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Alalem, M.; Ray, B.K. Loss of Epigenetic Kruppel-like Factor 4 Histone Deacetylase (KLF-4-HDAC)-mediated Transcriptional Suppression Is Crucial in Increasing Vascular Endothelial Growth Factor (VEGF) Expression in Breast Cancer. J. Biol. Chem. 2013, 288, 27232–27242. [Google Scholar] [CrossRef] [Green Version]
- Park, D.; Park, H.; Kim, Y.; Kim, H.; Jeoung, D. HDAC3 acts as a negative regulator of angiogenesis. BMB Rep. 2014, 47, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.; Huang, X.; Yang, Y.; Zhu, W.-G.; Gu, W.; Luo, J. Regulation of Histone Acetyltransferase TIP60 Function by Histone Deacetylase 3. J. Biol. Chem. 2014, 289, 33878–33886. [Google Scholar] [CrossRef] [Green Version]
- Deardorff, M.A.; Porter, N.J.; Christianson, D.W. Structural aspects of HDAC8 mechanism and dysfunction in Cornelia de Lange syndrome spectrum disorders: Structural Aspects of HDAC8 Mechanism. Protein Sci. 2016, 25, 1965–1976. [Google Scholar] [CrossRef] [Green Version]
- Geng, H.; Harvey, C.T.; Pittsenbarger, J.; Liu, Q.; Beer, T.M.; Xue, C.; Qian, D.Z. HDAC4 Protein Regulates HIF1α Protein Lysine Acetylation and Cancer Cell Response to Hypoxia. J. Biol. Chem. 2011, 286, 38095–38102. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.-C.; Cohen, T.J.; Barrientos, T.; Wang, B.; Li, M.; Simmons, B.J.; Yang, J.S.; Cox, G.A.; Zhao, Y.; Yao, T.-P. A Direct HDAC4-MAP Kinase Crosstalk Activates Muscle Atrophy Program. Mol. Cell 2012, 47, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Kaowinn, S.; Kaewpiboon, C.; Koh, S.; Krämer, O.; Chung, Y. STAT1‑HDAC4 signaling induces epithelial‑mesenchymal transition and sphere formation of cancer cells overexpressing the oncogene, CUG2. Oncol. Rep. 2018. [Google Scholar] [CrossRef]
- Mottet, D.; Pirotte, S.; Lamour, V.; Hagedorn, M.; Javerzat, S.; Bikfalvi, A.; Bellahcène, A.; Verdin, E.; Castronovo, V. HDAC4 represses p21WAF1/Cip1 expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene 2009, 28, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Dunaway, C.M.; Hwang, Y.; Lindsley, C.W.; Cook, R.S.; Wu, J.Y.; Boothby, M.; Chen, J.; Brantley-Sieders, D.M. Cooperative Signaling between Slit2 and Ephrin-A1 Regulates a Balance between Angiogenesis and Angiostasis. Mol. Cell. Biol. 2011, 31, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Urbich, C.; Rossig, L.; Kaluza, D.; Potente, M.; Boeckel, J.-N.; Knau, A.; Diehl, F.; Geng, J.-G.; Hofmann, W.-K.; Zeiher, A.M.; et al. HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood 2009, 113, 5669–5679. [Google Scholar] [CrossRef] [Green Version]
- Sucharov, C.C.; Dockstader, K.; McKinsey, T.A. YY1 Protects Cardiac Myocytes from Pathologic Hypertrophy by Interacting with HDAC5. Mol. Biol. Cell 2008, 19, 4141–4153. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Shor, A.C.; Bagui, T.K.; Seto, E.; Pledger, W.J. Histone deacetylase 5 represses the transcription of cyclin D3. J. Cell. Biochem. 2008, 104, 2143–2154. [Google Scholar] [CrossRef]
- Chen, S.; Yin, C.; Lao, T.; Liang, D.; He, D.; Wang, C.; Sang, N. AMPK-HDAC5 pathway facilitates nuclear accumulation of HIF-1α and functional activation of HIF-1 by deacetylating Hsp70 in the cytosol. Cell Cycle 2015, 14, 2520–2536. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Sun, S.; Yao, S.; Han, X.; Gu, M.; Shi, J. Histone deacetylase 5 promotes the proliferation and invasion of lung cancer cells. Oncol. Rep. 2018. [Google Scholar] [CrossRef] [Green Version]
- Raichur, S.; Hooi Teh, S.; Ohwaki, K.; Gaur, V.; Chau Long, Y.; Hargreaves, M.; McGee, S.L.; Kusunoki, J. Histone deacetylase 5 regulates glucose uptake and insulin action in muscle cells. J. Mol. Endocrinol. 2012, 49, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Dressel, U.; Bailey, P.J.; Wang, S.-C.M.; Downes, M.; Evans, R.M.; Muscat, G.E.O. A Dynamic Role for HDAC7 in MEF2-mediated Muscle Differentiation. J. Biol. Chem. 2001, 276, 17007–17013. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Liu, L.; Zhang, S.; Guo, S.; Li, X.; Wang, J.; Su, B.; Fang, Y.; Chen, X.; Ke, H.; et al. Hdac7 promotes lung tumorigenesis by inhibiting Stat3 activation. Mol. Cancer 2017, 16, 170. [Google Scholar] [CrossRef] [Green Version]
- Barneda-Zahonero, B.; Román-González, L.; Collazo, O.; Rafati, H.; Islam, A.B.M.M.K.; Bussmann, L.H.; di Tullio, A.; De Andres, L.; Graf, T.; López-Bigas, N.; et al. HDAC7 Is a Repressor of Myeloid Genes Whose Downregulation Is Required for Transdifferentiation of Pre-B Cells into Macrophages. PLoS Genet. 2013, 9, e1003503. [Google Scholar] [CrossRef] [Green Version]
- Witt, A.E.; Lee, C.-W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 2017, 36, 1707–1720. [Google Scholar] [CrossRef]
- Yuan, Z.; Peng, L.; Radhakrishnan, R.; Seto, E. Histone Deacetylase 9 (HDAC9) Regulates the Functions of the ATDC (TRIM29) Protein. J. Biol. Chem. 2010, 285, 39329–39338. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, Z.; Wang, N.; Guo, M.; Chi, X.; Pan, Y.; Jiang, J.; Niu, J.; Ksimu, S.; Li, J.Z.; et al. Role of HDAC9-FoxO1 Axis in the Transcriptional Program Associated with Hepatic Gluconeogenesis. Sci. Rep. 2017, 7, 6102. [Google Scholar] [CrossRef] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.-F.; Yao, T.-P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Scroggins, B.T.; Robzyk, K.; Wang, D.; Marcu, M.G.; Tsutsumi, S.; Beebe, K.; Cotter, R.J.; Felts, S.; Toft, D.; Karnitz, L.; et al. An Acetylation Site in the Middle Domain of Hsp90 Regulates Chaperone Function. Mol. Cell 2007, 25, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.-P. The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Ridinger, J.; Koeneke, E.; Kolbinger, F.R.; Koerholz, K.; Mahboobi, S.; Hellweg, L.; Gunkel, N.; Miller, A.K.; Peterziel, H.; Schmezer, P.; et al. Dual role of HDAC10 in lysosomal exocytosis and DNA repair promotes neuroblastoma chemoresistance. Sci. Rep. 2018, 8, 10039. [Google Scholar] [CrossRef] [Green Version]
- Duan, B.; Ye, D.; Zhu, S.; Jia, W.; Lu, C.; Wang, G.; Guo, X.; Yu, Y.; Wu, C.; Kang, J. HDAC10 promotes angiogenesis in endothelial cells through the PTPN22/ERK axis. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Shinsky, S.A.; Porter, N.J.; Christianson, D.W. Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat. Commun. 2017, 8, 15368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, R.; Li, Y.; Xiang, S.; Yuan, F.; Yuan, Z.; Telles, E.; Fang, J.; Coppola, D.; Shibata, D.; Lane, W.S.; et al. Histone Deacetylase 10 Regulates DNA Mismatch Repair and May Involve the Deacetylation of MutS Homolog 2. J. Biol. Chem. 2015, 290, 22795–22804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deubzer, H.E.; Schier, M.C.; Oehme, I.; Lodrini, M.; Haendler, B.; Sommer, A.; Witt, O. HDAC11 is a novel drug target in carcinomas. Int. J. Cancer 2013, 132, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Waltregny, D.; North, B.; Van Mellaert, F.; de Leval, J.; Verdin, E.; Castronovo, V. Screening of histone deacetylases (HDAC) expression in human prostate cancer reveals distinct class I HDAC profiles between epithelial and stromal cells. Eur. J. Histochem. EJH 2004, 48, 273–290. [Google Scholar] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Bertrand, P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [Google Scholar] [CrossRef]
- Robey, R.W.; Chakraborty, A.R.; Basseville, A.; Luchenko, V.; Bahr, J.; Zhan, Z.; Bates, S.E. Histone Deacetylase Inhibitors: Emerging Mechanisms of Resistance. Mol. Pharm. 2011, 8, 2021–2031. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. The Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Navakauskienė, R. Combination Epigenetic Therapy. In Handbook of Epigenetics; Elsevier, 2017; pp. 623–632. ISBN 978-0-12-805388-1. [Google Scholar]
- Bradley, D.; Rathkopf, D.; Dunn, R.; Stadler, W.M.; Liu, G.; Smith, D.C.; Pili, R.; Zwiebel, J.; Scher, H.; Hussain, M. Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): Trial results and interleukin-6 analysis: A study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 Consortium. Cancer 2009, 115, 5541–5549. [Google Scholar]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Yeruva, S.L.H.; Zhao, F.; Miller, K.D.; Tevaarwerk, A.J.; Wagner, L.I.; Gray, R.J.; Sparano, J.A.; Connolly, R.M. E2112: Randomized phase iii trial of endocrine therapy plus entinostat/placebo in patients with hormone receptor-positive advanced breast cancer. Npj Breast Cancer 2018, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yardley, D.A.; Ismail-Khan, R.R.; Melichar, B.; Lichinitser, M.; Munster, P.N.; Klein, P.M.; Cruickshank, S.; Miller, K.D.; Lee, M.J.; Trepel, J.B. Randomized Phase II, Double-Blind, Placebo-Controlled Study of Exemestane With or Without Entinostat in Postmenopausal Women With Locally Recurrent or Metastatic Estrogen Receptor-Positive Breast Cancer Progressing on Treatment With a Nonsteroidal Aromatase Inhibitor. J. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar] [PubMed] [Green Version]
- Jespersen, H.; Olofsson Bagge, R.; Ullenhag, G.; Carneiro, A.; Helgadottir, H.; Ljuslinder, I.; Levin, M.; All-Eriksson, C.; Andersson, B.; Stierner, U.; et al. Concomitant use of pembrolizumab and entinostat in adult patients with metastatic uveal melanoma (PEMDAC study): Protocol for a multicenter phase II open label study. BMC Cancer 2019, 19, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, J.E.; Saltos, A.; Tanvetyanon, T.; Haura, E.B.; Creelan, B.; Antonia, S.J.; Shafique, M.; Zheng, H.; Dai, W.; Saller, J.J.; et al. Phase I/Ib Study of Pembrolizumab Plus Vorinostat in Advanced/Metastatic Non–Small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 6623–6632. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.Z.; Wei, Y.-F.; Wang, X.; Kato, Y.; Cheng, L.; Pili, R. Antitumor activity of the histone deacetylase inhibitor MS-275 in prostate cancer models. The Prostate 2007, 67, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Boumber, Y.; Younes, A.; Garcia-Manero, G. Mocetinostat (MGCD0103): A review of an isotype-specific histone deacetylase inhibitor. Expert Opin. Investig. Drugs 2011, 20, 823–829. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.; Tse, E.; Kwong, Y.-L. Chidamide in the treatment of peripheral T-cell lymphoma. OncoTargets Ther. 2017, 10, 347–352. [Google Scholar] [CrossRef] [Green Version]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. In Advances in Cancer Research; Elsevier, 2018; Volume 138, pp. 183–211. ISBN 978-0-12-815127-3. [Google Scholar]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.-H.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar]
- Novotny-Diermayr, V.; Sangthongpitag, K.; Hu, C.Y.; Wu, X.; Sausgruber, N.; Yeo, P.; Greicius, G.; Pettersson, S.; Liang, A.L.; Loh, Y.K.; et al. SB939, a Novel Potent and Orally Active Histone Deacetylase Inhibitor with High Tumor Exposure and Efficacy in Mouse Models of Colorectal Cancer. Mol. Cancer Ther. 2010, 9, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Fournel, M.; Bonfils, C.; Hou, Y.; Yan, P.T.; Trachy-Bourget, M.-C.; Kalita, A.; Liu, J.; Lu, A.-H.; Zhou, N.Z.; Robert, M.-F.; et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 759–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, L.M.; Agus, D.B.; Scher, H.I.; Higgins, B.; Rose, A.; Cordon-Cardo, C.; Thaler, H.T.; Rifkind, R.A.; Marks, P.A.; Richon, V.M. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000, 60, 5165–5170. [Google Scholar] [PubMed]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Hussein, M.; Belani, C.; Robert, F.; Galanis, E.; Richon, V.M.; Garcia-Vargas, J.; Sanz-Rodriguez, C.; Rizvi, S. Vorinostat in solid and hematologic malignancies. J. Hematol. Oncol. 2009, 2, 31. [Google Scholar] [CrossRef] [Green Version]
- Siegel, D.S.; Hussein, M.A.; Belani, C.P.; Robert, F.; Rizvi, S.; Wigginton, J.; Randolph, S.; Crane, R.; Ganesan, R.; Garcia-Vargas, J. Safety and tolerability of vorinostat—Experience from the vorinostat clinical trial program. J. Clin. Oncol. 2008, 26, 14580. [Google Scholar] [CrossRef]
- Woods Ignatoski, K.M.; Friedman, J.; Escara-Wilke, J.; Zhang, X.; Daignault, S.; Dunn, R.L.; Smith, D.C.; Keller, E.T. Change in Markers of Bone Metabolism with Chemotherapy for Advanced Prostate Cancer: Interleukin-6 Response Is a Potential Early Indicator of Response to Therapy. J. Interferon Cytokine Res. 2009, 29, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Raedler, L.A. Farydak (Panobinostat): First HDAC Inhibitor Approved for Patients with Relapsed Multiple Myeloma. Am. Health Drug Benefits 2016, 9, 84–87. [Google Scholar]
- Chuang, M.-J.; Wu, S.-T.; Tang, S.-H.; Lai, X.-M.; Lai, H.-C.; Hsu, K.-H.; Sun, K.-H.; Sun, G.-H.; Chang, S.-Y.; Yu, D.-S.; et al. The HDAC Inhibitor LBH589 Induces ERK-Dependent Prometaphase Arrest in Prostate Cancer via HDAC6 Inactivation and Down-Regulation. PLoS ONE 2013, 8, e73401. [Google Scholar] [CrossRef]
- Welsbie, D.S.; Xu, J.; Chen, Y.; Borsu, L.; Scher, H.I.; Rosen, N.; Sawyers, C.L. Histone Deacetylases Are Required for Androgen Receptor Function in Hormone-Sensitive and Castrate-Resistant Prostate Cancer. Cancer Res. 2009, 69, 958–966. [Google Scholar] [CrossRef] [Green Version]
- Rathkopf, D.; Wong, B.Y.; Ross, R.W.; Anand, A.; Tanaka, E.; Woo, M.M.; Hu, J.; Dzik-Jurasz, A.; Yang, W.; Scher, H.I. A phase I study of oral panobinostat alone and in combination with docetaxel in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2010, 66, 181–189. [Google Scholar] [CrossRef]
- Molife, L.R.; Attard, G.; Fong, P.C.; Karavasilis, V.; Reid, A.H.M.; Patterson, S.; Riggs, C.E.; Higano, C.; Stadler, W.M.; McCulloch, W.; et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann. Oncol. 2010, 21, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Barbarotta, L.; Hurley, K. Romidepsin for the Treatment of Peripheral T-Cell Lymphoma. J. Adv. Pract. Oncol. 2015, 6, 22–36. [Google Scholar] [PubMed]
- de Bono, J.S.; Kristeleit, R.; Tolcher, A.; Fong, P.; Pacey, S.; Karavasilis, V.; Mita, M.; Shaw, H.; Workman, P.; Kaye, S.; et al. Phase I Pharmacokinetic and Pharmacodynamic Study of LAQ824, a Hydroxamate Histone Deacetylase Inhibitor with a Heat Shock Protein-90 Inhibitory Profile, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2008, 14, 6663–6673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solit, D. Hsp90 as a therapeutic target in prostate cancer. Semin. Oncol. 2003, 30, 709–716. [Google Scholar] [CrossRef]
- Razak, A.R.A.; Hotte, S.J.; Siu, L.L.; Chen, E.X.; Hirte, H.W.; Powers, J.; Walsh, W.; Stayner, L.-A.; Laughlin, A.; Novotny-Diermayr, V.; et al. Phase I clinical, pharmacokinetic and pharmacodynamic study of SB939, an oral histone deacetylase (HDAC) inhibitor, in patients with advanced solid tumours. Br. J. Cancer 2011, 104, 756–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eigl, B.J.; North, S.; Winquist, E.; Finch, D.; Wood, L.; Sridhar, S.S.; Powers, J.; Good, J.; Sharma, M.; Squire, J.A.; et al. A phase II study of the HDAC inhibitor SB939 in patients with castration resistant prostate cancer: NCIC clinical trials group study IND195. Invest. New Drugs 2015, 33, 969–976. [Google Scholar] [CrossRef]
- Halsall, J.A.; Turner, B.M. Histone deacetylase inhibitors for cancer therapy: An evolutionarily ancient resistance response may explain their limited success. BioEssays 2016, 38, 1102–1110. [Google Scholar] [CrossRef] [Green Version]
- Kantharaj, E.; Jayaram, R. Histone Deacetylase Inhibitors as Therapeutic Agents for Cancer Therapy: Drug Metabolism and Pharmacokinetic Properties. In Drug Development—A Case Study Based Insight into Modern Strategies; Rundfeldt, C., Ed.; InTech, 2011; ISBN 978-953-307-257-9. [Google Scholar]
- Konsoula, Z.; Cao, H.; Velena, A.; Jung, M. Pharmacokinetics-pharmacodynamics and antitumor activity of mercaptoacetamide-based histone deacetylase inhibitors. Mol. Cancer Ther. 2009, 8, 2844–2851. [Google Scholar] [CrossRef] [Green Version]
- Rundfeldt, C. Drug Development: A Case Study Based Insight into Modern Strategies; InTech: Rijeka, Croatia, 2011; ISBN 978-953-307-257-9. [Google Scholar]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Rathkopf, D.E.; Picus, J.; Hussain, A.; Ellard, S.; Chi, K.N.; Nydam, T.; Allen-Freda, E.; Mishra, K.K.; Porro, M.G.; Scher, H.I.; et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2013, 72, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leav, I.; Lau, K.M.; Adams, J.Y.; McNeal, J.E.; Taplin, M.E.; Wang, J.; Singh, H.; Ho, S.M. Comparative studies of the estrogen receptors beta and alpha and the androgen receptor in normal human prostate glands, dysplasia, and in primary and metastatic carcinoma. Am. J. Pathol. 2001, 159, 79–92. [Google Scholar] [CrossRef]
- Shah, R.B.; Mehra, R.; Chinnaiyan, A.M.; Shen, R.; Ghosh, D.; Zhou, M.; MacVicar, G.R.; Varambally, S.; Harwood, J.; Bismar, T.A.; et al. Androgen-Independent Prostate Cancer Is a Heterogeneous Group of Diseases: Lessons from a Rapid Autopsy Program. Cancer Res. 2004, 64, 9209–9216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scher, H.I.; Beer, T.M.; Higano, C.S.; Anand, A.; Taplin, M.-E.; Efstathiou, E.; Rathkopf, D.; Shelkey, J.; Yu, E.Y.; Alumkal, J.; et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: A phase 1–2 study. The Lancet 2010, 375, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Nickols, N.G.; Dervan, P.B. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc. Natl. Acad. Sci. USA 2007, 104, 10418–10423. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Coetzee, G.A. Prostate specific antigen gene regulation by androgen receptor. J. Cell. Biochem. 2004, 93, 233–241. [Google Scholar] [CrossRef]
- Saxena, P.; Trerotola, M.; Wang, T.; Li, J.; Sayeed, A.; VanOudenhove, J.; Adams, D.S.; FitzGerald, T.J.; Altieri, D.C.; Languino, L.R. PSA regulates androgen receptor expression in prostate cancer cells. The Prostate 2012, 72, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Bernhart, E.; Stuendl, N.; Kaltenegger, H.; Windpassinger, C.; Donohue, N.; Leithner, A.; Lohberger, B. Histone deacetylase inhibitors vorinostat and panobinostat induce G1 cell cycle arrest and apoptosis in multidrug resistant sarcoma cell lines. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.B.; Ki, S.W.; Yosnida, M.; Horinouchi, S. Mechanism of Cell Cycle Arrest Caused by Histone Deacetylase Inhibitors in Human Carcinoma Cells. J. Antibiot. (Tokyo) 2000, 53, 1191–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, G.-M.; Wang, H.-S.; Zhang, F.; Zhang, K.-S.; Liu, Z.-C.; Fang, R.; Wang, H.; Cai, S.-H.; Du, J. Histone deacetylase inhibitor induction of epithelial–mesenchymal transitions via up-regulation of Snail facilitates cancer progression. Biochim. Biophys. Acta BBA - Mol. Cell Res. 2013, 1833, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Yu, D.; Wu, H.; Liu, H.; Zhou, H.; Gu, R.; Zhang, R.; Zhang, S.; Wu, G. Anticancer activity of SAHA, a potent histone deacetylase inhibitor, in NCI-H460 human large-cell lung carcinoma cells in vitro and in vivo. Int. J. Oncol. 2014, 44, 451–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Zhou, F.; Lin, Z.; Shi, L.; Huang, A.; Liu, T.; Yu, D.; Wu, G. Antitumor effects of histone deacetylase inhibitor suberoylanilide hydroxamic acid in epidermal growth factor receptor-mutant non-small-cell lung cancer lines in vitro and in vivo. Anticancer Drugs 2018, 29, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Bausch, D.; Knightly, T.; Liu, Z.; Li, Y.; Liu, B.; Lu, J.; Chong, W.; Velmahos, G.C.; Alam, H.B. Histone deacetylase inhibitors enhance endothelial cell sprouting angiogenesis in vitro. Surgery 2011, 150, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.-M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Aurora, A.B.; Biyashev, D.; Mirochnik, Y.; Zaichuk, T.A.; Sanchez-Martinez, C.; Renault, M.-A.; Losordo, D.; Volpert, O.V. NF- B balances vascular regression and angiogenesis via chromatin remodeling and NFAT displacement. Blood 2010, 116, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Turtoi, A.; Mottet, D.; Matheus, N.; Dumont, B.; Peixoto, P.; Hennequière, V.; Deroanne, C.; Colige, A.; De Pauw, E.; Bellahcène, A.; et al. The angiogenesis suppressor gene AKAP12 is under the epigenetic control of HDAC7 in endothelial cells. Angiogenesis 2012, 15, 543–554. [Google Scholar] [CrossRef]
- Robey, R.W. Increased MDR1 Expression in Normal and Malignant Peripheral Blood Mononuclear Cells Obtained from Patients Receiving Depsipeptide (FR901228, FK228, NSC630176). Clin. Cancer Res. 2006, 12, 1547–1555. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.-F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 2008, 38, 802–832. [Google Scholar] [CrossRef]
- Mickley, L.A.; Bates, S.E.; Richert, N.D.; Currier, S.; Tanaka, S.; Foss, F.; Rosen, N.; Fojo, A.T. Modulation of the expression of a multidrug resistance gene (mdr-1/P-glycoprotein) by differentiating agents. J. Biol. Chem. 1989, 264, 18031–18040. [Google Scholar]
- Wang, H.; Huang, C.; Zhao, L.; Zhang, H.; Yang, J.M.; Luo, P.; Zhan, B.-X.; Pan, Q.; Li, J.; Wang, B.-L. Histone deacetylase inhibitors regulate P-gp expression in colorectal cancer via transcriptional activation and mRNA stabilization. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Yagüe, E.; Armesilla, A.L.; Harrison, G.; Elliott, J.; Sardini, A.; Higgins, C.F.; Raguz, S. P-glycoprotein (MDR1) Expression in Leukemic Cells Is Regulated at Two Distinct Steps, mRNA Stabilization and Translational Initiation. J. Biol. Chem. 2003, 278, 10344–10352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, H.; Wang, C.; Zhou, K.; Wang, T.; Li, Y.; Qiu, D.; Li, Q.; Zhang, Y.; Hua, Y. The effect of histone deacetylase inhibition on the expression of P-glycoprotein in human placental trophoblast cell lines. Placenta 2017, 49, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Noack, A.; Noack, S.; Buettner, M.; Naim, H.Y.; Löscher, W. Intercellular transfer of P-glycoprotein in human blood-brain barrier endothelial cells is increased by histone deacetylase inhibitors. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Shukla, S.; Ohnuma, S.; Ambudkar, S.V. Improving cancer chemotherapy with modulators of ABC drug transporters. Curr. Drug Targets 2011, 12, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.J. Chemoresistance to Depsipeptide FK228 [(E)-(1S,4S,10S,21R)-7-[(Z)-Ethylidene]-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,20,23-tetraazabicyclo[8,7,6]-tricos-16-ene-3,6,9,22-pentanone] Is Mediated by Reversible MDR1 Induction in Human Cancer Cell Lines. J. Pharmacol. Exp. Ther. 2005, 314, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Ruefli, A.A.; Bernhard, D.; Tainton, K.M.; Kofler, R.; Smyth, M.J.; Johnstone, R.W. Suberoylanilide hydroxamic acid (SAHA) overcomes multidrug resistance and induces cell death in P-glycoprotein-expressing cells. Int. J. Cancer 2002, 99, 292–298. [Google Scholar] [CrossRef]
- Sánchez, C.; Mendoza, P.; Contreras, H.R.; Vergara, J.; McCubrey, J.A.; Huidobro, C.; Castellón, E.A. Expression of multidrug resistance proteins in prostate cancer is related with cell sensitivity to chemotherapeutic drugs. The Prostate 2009, 69, 1448–1459. [Google Scholar] [CrossRef]
- Fiskus, W.; Rao, R.; Fernandez, P.; Herger, B.; Yang, Y.; Chen, J.; Kolhe, R.; Mandawat, A.; Wang, Y.; Joshi, R.; et al. Molecular and biologic characterization and drug sensitivity of pan-histone deacetylase inhibitor-resistant acute myeloid leukemia cells. Blood 2008, 112, 2896–2905. [Google Scholar] [CrossRef] [Green Version]
- Halsall, J.A.; Turan, N.; Wiersma, M.; Turner, B.M. Cells adapt to the epigenomic disruption caused by histone deacetylase inhibitors through a coordinated, chromatin-mediated transcriptional response. Epigenetics Chromatin 2015, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Lamparter, C.L.; Winn, L.M. Valproic acid exposure decreases Cbp/p300 protein expression and histone acetyltransferase activity in P19 cells. Toxicol. Appl. Pharmacol. 2016, 306, 69–78. [Google Scholar] [CrossRef]
- Alt, J.R.; Gladden, A.B.; Diehl, J.A. p21 Cip1 Promotes Cyclin D1 Nuclear Accumulation via Direct Inhibition of Nuclear Export. J. Biol. Chem. 2002, 277, 8517–8523. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Raina, K.; Agarwal, R. Deletion of p21/Cdkn1a confers protective effect against prostate tumorigenesis in transgenic adenocarcinoma of the mouse prostate model. Cell Cycle 2013, 12, 1598–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, B.-D.; Watanabe, K.; Broude, E.V.; Fang, J.; Poole, J.C.; Kalinichenko, T.V.; Roninson, I.B. Effects of p21Waf1/Cip1/Sdi1 on cellular gene expression: Implications for carcinogenesis, senescence, and age-related diseases. Proc. Natl. Acad. Sci. USA 2000, 97, 4291–4296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamloo, B.; Usluer, S. p21 in Cancer Research. Cancers 2019, 11, 1178. [Google Scholar] [CrossRef] [Green Version]
- Vrana, J.A.; Decker, R.H.; Johnson, C.R.; Wang, Z.; Jarvis, W.D.; Richon, V.M.; Ehinger, M.; Fisher, P.B.; Grant, S. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene 1999, 18, 7016–7025. [Google Scholar] [CrossRef] [Green Version]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagosklonny, M.V.; Bates, S.E. P21-dependent G1arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [Green Version]
- Negmeldin, A.T.; Knoff, J.R.; Pflum, M.K.H. The structural requirements of histone deacetylase inhibitors: C4-modified SAHA analogs display dual HDAC6/HDAC8 selectivity. Eur. J. Med. Chem. 2018, 143, 1790–1806. [Google Scholar] [CrossRef] [PubMed]
- Heider, U.; Rademacher, J.; Lamottke, B.; Mieth, M.; Moebs, M.; von Metzler, I.; Assaf, C.; Sezer, O. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in cutaneous T cell lymphoma. Eur. J. Haematol. 2009, 82, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc in Cellular Regulation: The Nature and Significance of “Zinc Signals”. Int. J. Mol. Sci. 2017, 18, 2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzellotti, A.I.; Farrell, N.P. Zinc metalloproteins as medicinal targets. Chem. Soc. Rev. 2008, 37, 1629. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HDAC Class | HDAC Type | Localization | Function |
|---|---|---|---|
| I | 1 /2 | Nuclear | Interact with the promoters of vimentin, Slug and ZEB1 and upregulate EMT [22] Promotes DNA repair via NHEJ [23] Significant in stem cell functionality and clonogenic capacity in intestinal and embryonic stem cells [24,25] HDAC2, in complex with HDAC3 and KLF4 reduces VEGF expression, downregulating angiogenesis [26] |
| 3 | Negative regulator of angiogenesis [27] TIP60 ubiquitination and cytoplasmic localization and protects cells from apoptosis after DNA damage [28] | ||
| 8 | Involved in deacetylation of SMC3 leading to cohesion recycling during the cell cycle [29] | ||
| IIa | 4 | Nuclear/ cytoplasmic | Downregulation results in HIF-1α, MEKK2 and STAT1 acetylation [30,31,32] Induction of p21 inhibited by HDAC4 silencing allowing cells to progress from G1 to S phase [33] |
| 5 | Interacts with the promoter of FGF2 and SLIT2, downregulating endothelial cell growth [34] Represses angiogenesis by downregulating FGF2 and Slit2, resulting in reduced sprout formation [35] Colocalizes with YY1, maintaining a terminally-differentiated state in cardiomyocyte cells (H92C) [36] Represses transcription of cyclin D3 [37] Required for nuclear accumulation of HIF-1α in hypoxic conditions [38] Promotes proliferation of tumor cells [39] Leads to reduced glucose uptake in primary muscle cells (with reduction in GLUT4 (SLC2A4) gene but increased GLUT1 expression) and insulin-stimulated glycogen synthesis [40] | ||
| 7 | Upregulates PDGF-B, negatively regulating angiogenesis Represses MEF2 activity [41] Inhibits Stat3 activity by directly deacetylating Stat3, promoting lung tumorigenesis [42] Represses myeloid genes, preventing transdifferentiation of pre-B cells into macrophages [43] Augments the CSC phenotype of breast and ovarian cancer [44] | ||
| 9 | Deacetylates ATDC, reducing ATDC-p53 interaction, and consequently inhibiting cell proliferation [45] Triggers gluconeogenesis by deacetylating FoxO1 and altering gene expression profiles of PGC-1α, cyclic AMP-responsive element-binding protein (CREB) and glucocorticoid receptor [46] | ||
| IIb | 6 | Nuclear/ cytoplasmic | Overexpression of HDAC6 in mammalian cells promotes chemotactic cell movement [47] Deacetylates HSP90, a chaperone protein, prolonging substrate protein action (e.g., AR) [48] Directly interacts with ubiquitin protein and dynein and transports misfolded proteins through microtubules to the perinuclear aggresomes. Polymers then degrade misfolded proteins by autophagy [49] |
| 10 | Involved in DNA double-strand break repair in neuroblastoma cells [50] Promotes angiogenesis by deacetylating PTPN22 in endothelial cells [51] Functions as a polyamine deacetylase [52] Overexpression in HeLa cells triggers cellular DNA MMR activity specifically by deacetylating MSH2 [53] | ||
| IV | 11 | Inhibition results in a differential expression of genes involved in cytoskeleton remodeling, chromatin assembly and transcription, particularly WNT and PPAR-signaling pathways. Regulates survival genes in colorectal cancer patient (HMOX1), growth inhibition (GCNT3), apoptosis (AK2, TFAP2A) and differentiation of colorectal cancer stem cells (BMP4) [54] |
| HDAC Inhibitor | Drug Combination | Tumor Type | Trial Phase Completed [Reference] |
|---|---|---|---|
| Entinostat | Exesmestane | Recurrent hormone receptor-positive breast cancer | Phase II trial [64] |
| Permolizumab | Metastatic uveal melanoma | Phase II trial [65] | |
| Chidamide | Exesmestane | Hormone receptor-positive, HER2-negative breast cancer | Phase III trial [62] |
| Vorinostat | Permolizumab | Advanced/metastatic Non-small cell lung cancer | Phase I trial [66] |
| Compound | HDAC Inhibition IC50 (nM) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Class I | Class IIa | Class IIb | Class IV | ||||||||
| 1 | 2 | 3 | 8 | 4 | 5 | 7 | 9 | 6 | 10 | 11 | |
| SAHA | 38 | 144 | 6 | 38 | >30000 | >30000 | >300000 | >30000 | 10 | 21 | 28 |
| Panobinostat | 3 | 3 | 4 | 248 | 23 | NA | 18 | 6 | 3 | ND | ND |
| Belinostat | 41 | 125 | 30 | 216 | 115 | NA | 67 | 128 | 82 | ND | ND |
| Pracinostat | 49 | 96 | 43 | 140 | 56 | 47 | 137 | 70 | 1008 | 40 | ND |
| Chidamide | 95 | 160 | 67 | 733 | >30000 | >30000 | >30000 | >30000 | >30000 | 78 | 432 |
| Entinostat | 262 | 306 | 499 | 2700 | >30000 | >30000 | >30000 | >30000 | >30000 | 254 | 0.649 |
| Mocetinostat | 150 | 290 | 1660 | >10000 | >10000 | >10000 | >10000 | ND | ND | ND | 590 |
| Romidepsin | 36 | 47 | ND | ND | 510 | ND | ND | ND | 14000 | ND | ND |
| Name | Structural Class | HDACs Inhibited | Prostate Cancer Clinical Trial Status |
|---|---|---|---|
| SAHA | Hydroxamic acid | I, IIb and IV | II failed |
| Depsipeptide (Romidepsin) | Cyclic peptide | HDAC 1, 2 and 4 | II failed |
| Pracinostat (SB939) | Hydroxamic acid | I, IIa and HDAC10 | II failed |
| Panobinostat | Hydroxamic acid | HDAC I, 4, 6, 7 and 9 | II failed |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer. Biomedicines 2020, 8, 22. https://doi.org/10.3390/biomedicines8020022
Rana Z, Diermeier S, Hanif M, Rosengren RJ. Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer. Biomedicines. 2020; 8(2):22. https://doi.org/10.3390/biomedicines8020022
Chicago/Turabian StyleRana, Zohaib, Sarah Diermeier, Muhammad Hanif, and Rhonda J. Rosengren. 2020. "Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer" Biomedicines 8, no. 2: 22. https://doi.org/10.3390/biomedicines8020022