Functional Characterization of Colon-Cancer-Associated Variants in ADAM17 Affecting the Catalytic Domain

 , , , and
, , , and
Abstract
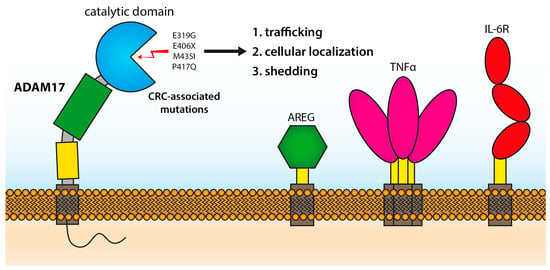
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
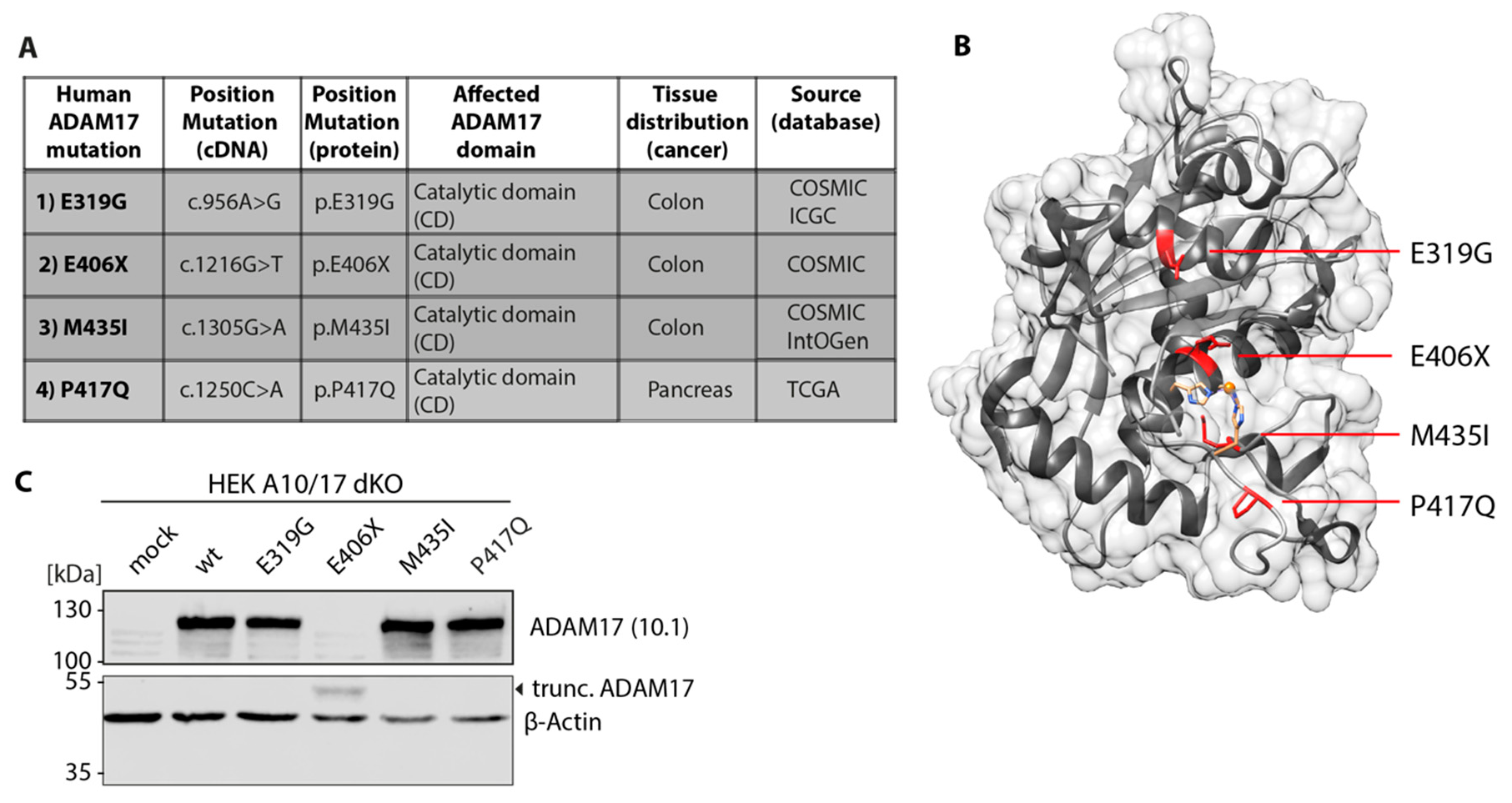
2.1. Database Analysis
2.2. cDNA Constructs and Cloning
2.3. Cell Culture
2.4. Transfection
2.5. Western Blot Analysis
2.6. Biotinylation
2.7. Immunofluorescence Analysis
2.8. Enzyme-Linked Immunosorbent Assay (ELISA)
2.9. Live Cell Surface ADAM17 Activity Assay
2.10. Structural Analysis
2.11. Data Analysis and Statistic
3. Results
3.1. Cloning and Expression of ADAM17 Mutations
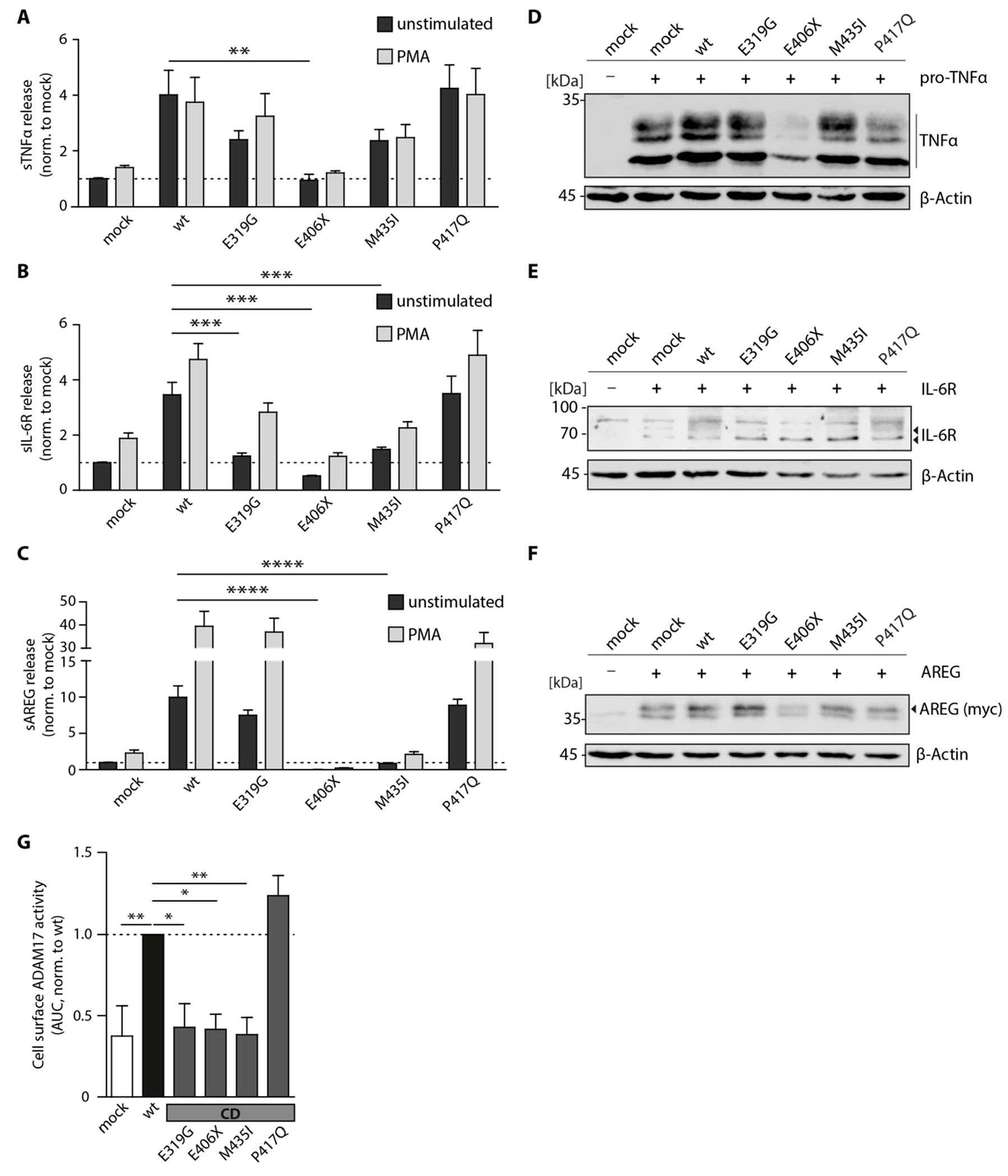
3.2. Proteolytic Activity of Cancer-Associated ADAM17 Variants
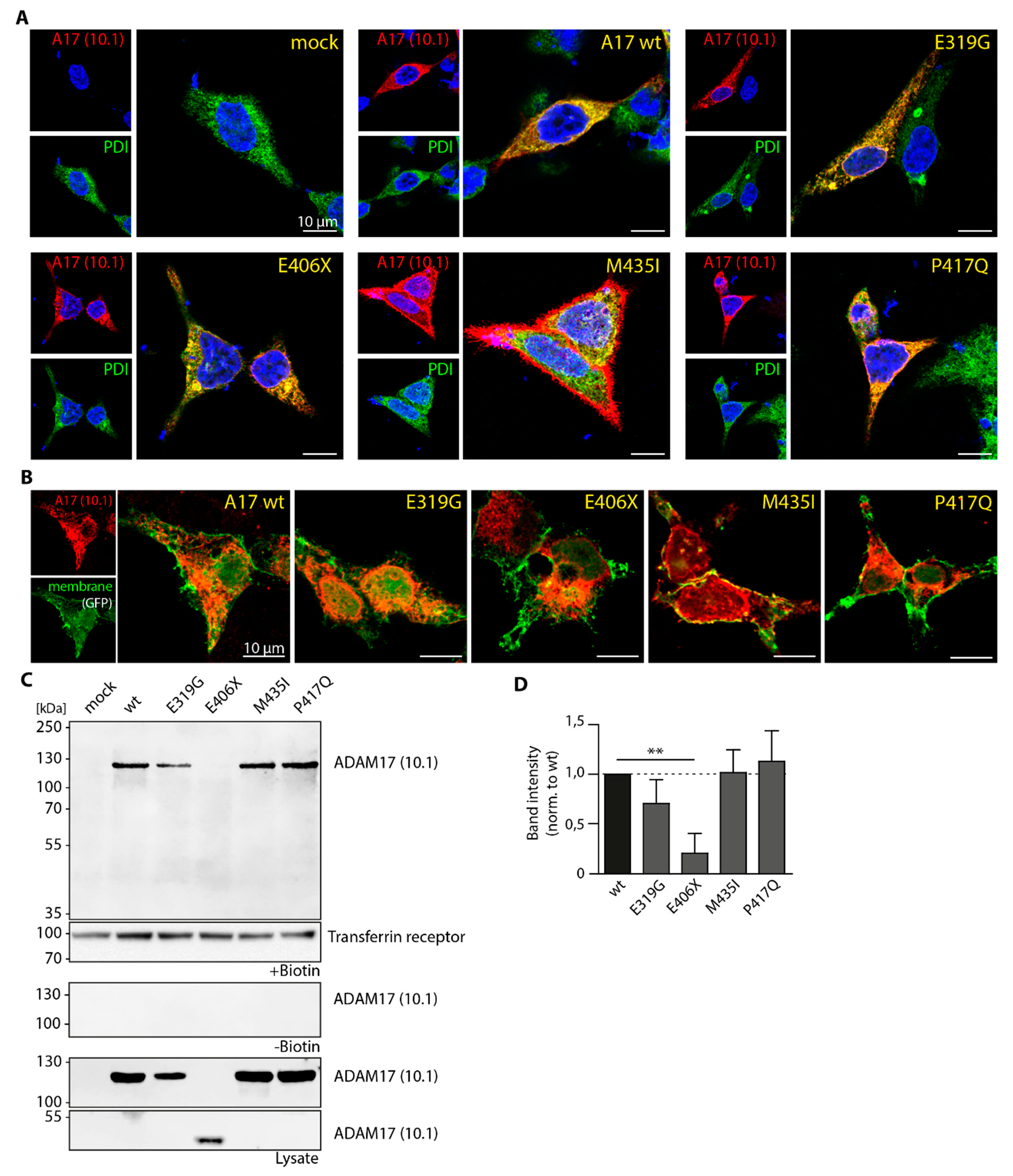
3.3. Cellular Localization of ADAM17 Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM17 | A Disintegrin and Metalloproteinase 17 |
| AREG | Amphiregulin |
| CD | catalytic domain |
| CRC | colorectal cancer |
| dKO | double knock-out |
| EGF-R | epidermal growth factor receptor |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| ER | endoplasmatic reticulum |
| HEK | human embryonic kidney cells |
| IL-6R | interleukin-6 receptor |
| kDa | kilo Dalton |
| m | murine |
| PDI | protein disulfide isomerase |
| PMA | phorbol 12-myristate 13-acetate |
| sTNFα | soluble tumor necrosis factor alpha |
| wt | wild type |
References
- Cancer Facts & Figures 2019. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019.html (accessed on 23 March 2020).
- Johnson, C.M.; Wei, C.; Ensor, J.E.; Smolenski, D.J.; Amos, C.I.; Levin, B.; Berry, D.A. Meta-analyses of colorectal cancer risk factors. Cancer Causes Control 2013, 24, 1207–1222. [Google Scholar] [CrossRef] [PubMed]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e2105. [Google Scholar] [CrossRef] [PubMed]
- Ponder, A.; Long, M.D. A clinical review of recent findings in the epidemiology of inflammatory bowel disease. Clin. Epidemiol. 2013, 5, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Pang, Z.; Chen, W.; Ju, S.; Zhou, C. The epidemiology and risk factors of inflammatory bowel disease. Int. J. Clin. Exp. Med. 2015, 8, 22529–22542. [Google Scholar] [PubMed]
- Amre, D.K.; D’Souza, S.; Morgan, K.; Seidman, G.; Lambrette, P.; Grimard, G.; Israel, D.; Mack, D.; Ghadirian, P.; Deslandres, C.; et al. Imbalances in dietary consumption of fatty acids, vegetables, and fruits are associated with risk for Crohn’s disease in children. Am. J. Gastroenterol. 2007, 102, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Lauterbach, M.; Latz, E. Western Diet and the Immune System: An Inflammatory Connection. Immunity 2019, 51, 794–811. [Google Scholar] [CrossRef]
- O’Neill, A.M.; Burrington, C.M.; Gillaspie, E.A.; Lynch, D.T.; Horsman, M.J.; Greene, M.W. High-fat Western diet-induced obesity contributes to increased tumor growth in mouse models of human colon cancer. Nutr. Res. 2016, 36, 1325–1334. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys Acta Mol. Cell Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef]
- Chalaris, A.; Adam, N.; Sina, C.; Rosenstiel, P.; Lehmann-Koch, J.; Schirmacher, P.; Hartmann, D.; Cichy, J.; Gavrilova, O.; Schreiber, S.; et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010, 207, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Gooz, M. ADAM-17: the enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 146–169. [Google Scholar] [CrossRef] [Green Version]
- Pavlenko, E.; Cabron, A.-S.; Arnold, P.; Dobert, J.; Rose-John, S.; Zunke, F. Functional Characterization of Colon Cancer-Associated Mutations in ADAM17: Modifications in the Pro-Domain Interfere with Trafficking and Maturation. Int. J. Mol. Sci. 2019, 20, 2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Düsterhöft, S.; Michalek, M.; Kordowski, F.; Oldefest, M.; Sommer, A.; Röseler, J.; Reiss, K.; Grötzinger, J.; Lorenzen, I. Extracellular Juxtamembrane Segment of ADAM17 Interacts with Membranes and Is Essential for Its Shedding Activity. Biochemistry 2015, 54, 5791–5801. [Google Scholar] [CrossRef] [PubMed]
- Peiretti, F.; Canault, M.; Deprez-Beauclair, P.; Berthet, V.; Bonardo, B.; Juhan-Vague, I.; Nalbone, G. Intracellular maturation and transport of tumor necrosis factor alpha converting enzyme. Exp. Cell Res. 2003, 285, 278–285. [Google Scholar] [CrossRef]
- Soond, S.M.; Everson, B.; Riches, D.W.; Murphy, G. ERK-mediated phosphorylation of Thr735 in TNFalpha-converting enzyme and its potential role in TACE protein trafficking. J. Cell Sci. 2005, 118, 2371–2380. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.; Schmidt, S.; Will, O.; Koudelka, T.; Kohler, K.; Boss, M.; Rabe, B.; Tholey, A.; Scheller, J.; Schmidt-Arras, D.; et al. Polo-like kinase 2, a novel ADAM17 signaling component, regulates tumor necrosis factor alpha ectodomain shedding. J. Biol. Chem. 2014, 289, 3080–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Derynck, R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell 2010, 37, 551–566. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Rodriguez, E.; Montero, J.C.; Esparis-Ogando, A.; Yuste, L.; Pandiella, A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor alpha-converting enzyme at threonine 735: A potential role in regulated shedding. Mol. Biol. Cell 2002, 13, 2031–2044. [Google Scholar] [CrossRef]
- Lorenzen, I.; Lokau, J.; Korpys, Y.; Oldefest, M.; Flynn, C.M.; Künzel, U.; Garbers, C.; Freeman, M.; Grötzinger, J.; Düsterhöft, S. Control of ADAM17 activity by regulation of its cellular localisation. Sci. Rep. 2016, 6, 35067. [Google Scholar] [CrossRef]
- Tellier, E.; Canault, M.; Rebsomen, L.; Bonardo, B.; Juhan-Vague, I.; Nalbone, G.; Peiretti, F. The shedding activity of ADAM17 is sequestered in lipid rafts. Exp. Cell Res. 2006, 312, 3969–3980. [Google Scholar] [CrossRef]
- Düsterhöft, S.; Jung, S.; Hung, C.W.; Tholey, A.; Sönnichsen, F.D.; Grötzinger, J.; Lorenzen, I. Membrane-proximal domain of a disintegrin and metalloprotease-17 represents the putative molecular switch of its shedding activity operated by protein-disulfide isomerase. J. Am. Chem. Soc. 2013, 135, 5776–5781. [Google Scholar] [CrossRef] [PubMed]
- Adrain, C.; Zettl, M.; Christova, Y.; Taylor, N.; Freeman, M. Tumor Necrosis Factor Signaling Requires iRhom2 to Promote Trafficking and Activation of TACE. Science 2012, 335, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Düsterhöft, S.; Babendreyer, A.; Giese, A.A.; Flasshove, C.; Ludwig, A. Status update on iRhom and ADAM17: It’s still complicated. Biochim Biophys Acta Mol. Cell Res. 2019, 1866, 1567–1583. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Lang, P.A.; Maretzky, T.; Hamada, K.; Ohishi, K.; Maney, S.K.; Berger, T.; Murthy, A.; Duncan, G.; Xu, H.C.; et al. iRhom2 Regulation of TACE Controls TNF-Mediated Protection Against Listeria and Responses to LPS. Science 2012, 335, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.; Li, X.; Maretzky, T.; Perez-Aguilar, J.M.; McIlwain, D.; Xie, Y.; Zheng, Y.; Mak, T.W.; Weinstein, H.; Blobel, C.P. Substrate-selective protein ectodomain shedding by ADAM17 and iRhom2 depends on their juxtamembrane and transmembrane domains. FASEB J. 2020, 34, 4956–4969. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, P.E.; Solomon, A.; Miller, A.B.; Leesnitzer, M.A.; Sagi, I.; Milla, M.E. Inhibition of the tumor necrosis factor-alpha-converting enzyme by its pro domain. J. Biol. Chem. 2004, 279, 31638–31645. [Google Scholar] [CrossRef] [Green Version]
- Mazzola, R.D., Jr.; Zhu, Z.; Sinning, L.; McKittrick, B.; Lavey, B.; Spitler, J.; Kozlowski, J.; Neng-Yang, S.; Zhou, G.; Guo, Z.; et al. Discovery of novel hydroxamates as highly potent tumor necrosis factor-alpha converting enzyme inhibitors. Part II: optimization of the S3’ pocket. Bioorg. Med. Chem. Lett. 2008, 18, 5809–5814. [Google Scholar] [CrossRef]
- Birkedal-Hansen, H. Role of cytokines and inflammatory mediators in tissue destruction. J. Periodontal Res. 1993, 28, 500–510. [Google Scholar] [CrossRef]
- Papadakis, K.A.; Targan, S.R. Role of Cytokines in the Pathogenesis of Inflammatory Bowel Disease. Annu. Rev. Med. 2000, 51, 289–298. [Google Scholar] [CrossRef]
- Garton, K.J.; Gough, P.J.; Blobel, C.P.; Murphy, G.; Greaves, D.R.; Dempsey, P.J.; Raines, E.W. Tumor Necrosis Factor-α-converting Enzyme (ADAM17) Mediates the Cleavage and Shedding of Fractalkine (CX3CL1). J. Biol. Chem. 2001, 276, 37993–38001. [Google Scholar] [PubMed]
- Garton, K.J.; Gough, P.J.; Philalay, J.; Wille, P.T.; Blobel, C.P.; Whitehead, R.H.; Dempsey, P.J.; Raines, E.W. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17). J. Biol. Chem. 2003, 278, 37459–37464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsakadze, N.L.; Sithu, S.D.; Sen, U.; English, W.R.; Murphy, G.; D’Souza, S.E. Tumor Necrosis Factor-α-converting Enzyme (TACE/ADAM-17) Mediates the Ectodomain Cleavage of Intercellular Adhesion Molecule-1 (ICAM-1). J. Biol. Chem. 2006, 281, 3157–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisi, S.; D’Amore, M.; Sisto, M. ADAM17 at the interface between inflammation and autoimmunity. Immunol. Lett. 2014, 162, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Müllberg, J.; Schooltink, H.; Stoyan, T.; Günther, M.; Graeve, L.; Buse, G.; Mackiewicz, A.; Heinrich, P.C.; Rose-John, S. The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 1993, 23, 473–480. [Google Scholar] [CrossRef]
- Wolf, J.; Rose-John, S.; Garbers, C. Interleukin-6 and its receptors: A highly regulated and dynamic system. Cytokine 2014, 70, 11–20. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2011, 1813, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, P.C.; Behrmann, I.; MÜLler-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway1. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [Green Version]
- Rose-John, S.; Heinrich, P.C. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem. J. 1994, 300 (Pt. 2), 281–290. [Google Scholar] [CrossRef]
- Chalaris, A.; Garbers, C.; Rabe, B.; Rose-John, S.; Scheller, J. The soluble Interleukin 6 receptor: Generation and role in inflammation and cancer. Eur. J. Cell Biol. 2011, 90, 484–494. [Google Scholar] [CrossRef]
- Rose-John, S. IL-6 Trans-Signaling via the Soluble IL-6 Receptor: Importance for the Pro-Inflammatory Activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, N.; Rose-John, S. ADAM17 Activity and IL-6 Trans-Signaling in Inflammation and Cancer. Cancers 2019, 11, 1736. [Google Scholar] [CrossRef] [Green Version]
- Blobel, C.P. ADAMs: key components in EGFR signalling and development. Nat. Rev. Mol. Cell. Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef]
- Sibilia, M.; Kroismayr, R.; Lichtenberger, B.M.; Natarajan, A.; Hecking, M.; Holcmann, M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation 2007, 75, 770–787. [Google Scholar] [CrossRef]
- Al Moustafa, A.E.; Achkhar, A.; Yasmeen, A. EGF-receptor signaling and epithelial-mesenchymal transition in human carcinomas. Front. Biosci (Sch. Ed.) 2012, 4, 671–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adriano, A.; Giovanni Luca, G.; Nadia, R.; Danilo, M.; Claudio, F.; Paola, M.; Anna, T.; Carlo, V.; Mauro, B. Suppression of EGF-R signaling reduces the incidence of prostate cancer metastasis in nude mice. Endocr. -Relat. Cancer Endocr. Relat. Cancer 2006, 13, 197–210. [Google Scholar] [CrossRef]
- Malecka-Panas, E.; Kordek, R.; Biernat, W.; Tureaud, J.; Liberski, P.P.; Majumdar, A.P. Differential activation of total and EGF receptor (EGF-R) tyrosine kinase (tyr-k) in the rectal mucosa in patients with adenomatous polyps, ulcerative colitis and colon cancer. Hepatogastroenterology 1997, 44, 435–440. [Google Scholar] [CrossRef]
- Berasain, C.; Castillo, J.; Prieto, J.; Avila, M.A. New molecular targets for hepatocellular carcinoma: the ErbB1 signaling system. Liver Int. 2007, 27, 174–185. [Google Scholar] [CrossRef]
- Blanchot-Jossic, F.; Jarry, A.; Masson, D.; Bach-Ngohou, K.; Paineau, J.; Denis, M.G.; Laboisse, C.L.; Mosnier, J.F. Up-regulated expression of ADAM17 in human colon carcinoma: co-expression with EGFR in neoplastic and endothelial cells. J. Pathol. 2005, 207, 156–163. [Google Scholar] [CrossRef]
- Schmidt, S.; Schumacher, N.; Schwarz, J.; Tangermann, S.; Kenner, L.; Schlederer, M.; Sibilia, M.; Linder, M.; Altendorf-Hofmann, A.; Knosel, T.; et al. ADAM17 is required for EGF-R-induced intestinal tumors via IL-6 trans-signaling. J. Exp. Med. 2018, 215, 1205–1225. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Fantini, M.C.; Wirtz, S.; Nikolaev, A.; Lehr, H.A.; Galle, P.R.; Rose-John, S.; Neurath, M.F. IL-6 signaling promotes tumor growth in colorectal cancer. Cell Cycle 2005, 4, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Hara, T.; Mitsuyama, K.; Yamamoto, M.; Tsuruta, O.; Sata, M.; Scheller, J.; Rose-John, S.; Kado, S.-i.; Takada, T. Essential Roles of IL-6 Trans-Signaling in Colonic Epithelial Cells, Induced by the IL-6/Soluble–IL-6 Receptor Derived from Lamina Propria Macrophages, on the Development of Colitis-Associated Premalignant Cancer in a Murine Model. J. Immunol. 2010, 184, 1543–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivatsa, S.; Paul, M.C.; Cardone, C.; Holcmann, M.; Amberg, N.; Pathria, P.; Diamanti, M.A.; Linder, M.; Timelthaler, G.; Dienes, H.P.; et al. EGFR in Tumor-Associated Myeloid Cells Promotes Development of Colorectal Cancer in Mice and Associates With Outcomes of Patients. Gastroenterology 2017, 153, 178–190.e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalogue of Somatic Mutations in Cancer (COSMIC). Available online: cancer.sanger.ac.uk (accessed on 27 February 2017).
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Integrative OncoGenomics (IntOGen). Available online: www.intogen.org (accessed on 27 February 2017).
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Program (TCGA). Available online: https://www.cancer.gov/tcga (accessed on 27 February 2017).
- International Cancer Genome, C.; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Pérez, L.; Kerrigan, J.E.; Li, X.; Fan, H. Substitution of methionine 435 with leucine, isoleucine, and serine in tumor necrosis factor alpha converting enzyme inactivates ectodomain shedding activity. Biochem. Cell. Biol. 2007, 85, 141–149. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Sanner, M.F.; Olson, A.J.; Spehner, J.C. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers 1996, 38, 305–320. [Google Scholar] [CrossRef]
- Cabron, A.-S.; El Azzouzi, K.; Boss, M.; Arnold, P.; Schwarz, J.; Rosas, M.; Dobert, J.P.; Pavlenko, E.; Schumacher, N.; Renné, T.; et al. Structural and Functional Analyses of the Shedding Protease ADAM17 in HoxB8-Immortalized Macrophages and Dendritic-like Cells. J. Immunol. 2018, 201, 3106–3118. [Google Scholar] [CrossRef] [Green Version]
- Saad, M.I.; Rose-John, S.; Jenkins, B.J. ADAM17: An Emerging Therapeutic Target for Lung Cancer. Cancers 2019, 11, 1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoseki, J.; Ushioda, R.; Nagata, K. Mechanism and components of endoplasmic reticulum-associated degradation. J. Biochem. 2009, 147, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobert, J.P.; Cabron, A.-S.; Arnold, P.; Pavlenko, E.; Rose-John, S.; Zunke, F. Functional Characterization of Colon-Cancer-Associated Variants in ADAM17 Affecting the Catalytic Domain. Biomedicines 2020, 8, 463. https://doi.org/10.3390/biomedicines8110463
Dobert JP, Cabron A-S, Arnold P, Pavlenko E, Rose-John S, Zunke F. Functional Characterization of Colon-Cancer-Associated Variants in ADAM17 Affecting the Catalytic Domain. Biomedicines. 2020; 8(11):463. https://doi.org/10.3390/biomedicines8110463
Chicago/Turabian StyleDobert, Jan Philipp, Anne-Sophie Cabron, Philipp Arnold, Egor Pavlenko, Stefan Rose-John, and Friederike Zunke. 2020. "Functional Characterization of Colon-Cancer-Associated Variants in ADAM17 Affecting the Catalytic Domain" Biomedicines 8, no. 11: 463. https://doi.org/10.3390/biomedicines8110463