Emerging Concepts and Challenges in Rheumatoid Arthritis Gene Therapy

 , and
, and
Abstract
:1. Introduction
2. Targets for RA Gene Therapy
2.1. Anti- and Proinflammatory Cytokines
2.2. Matrix Degradation Enzymes
2.3. Hormonal Regulation Proteins
2.4. Noncoding RNA Molecules
2.4.1. MicroRNAs
2.4.2. Long Noncoding RNAs
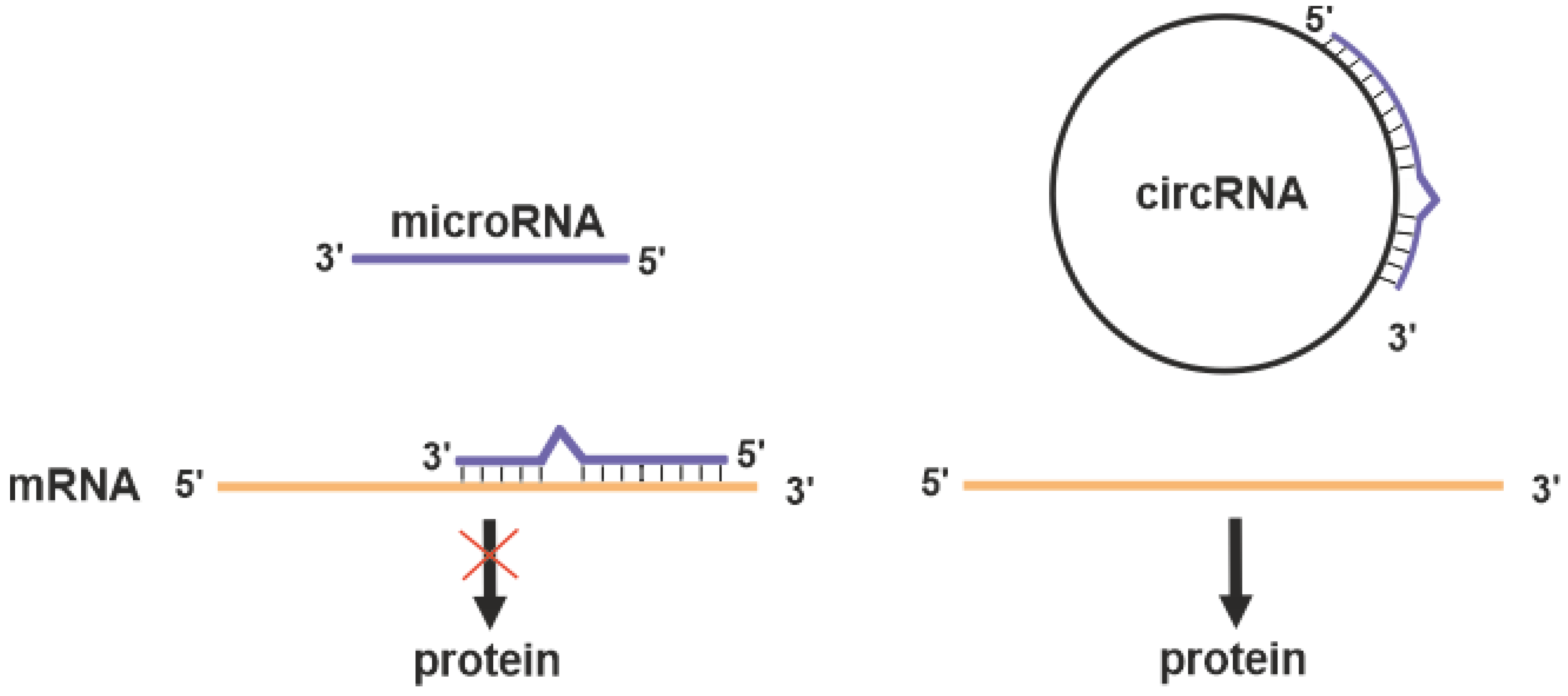
2.4.3. Circular RNAs
3. RA Gene Therapy Delivery
3.1. Local or Systemic Delivery
3.2. Ex Vivo or In Vivo
3.3. RNA Therapeutics
3.3.1. RNA Interference
3.3.2. Circular RNA
3.3.3. Nonviral RNA Delivery Vehicles
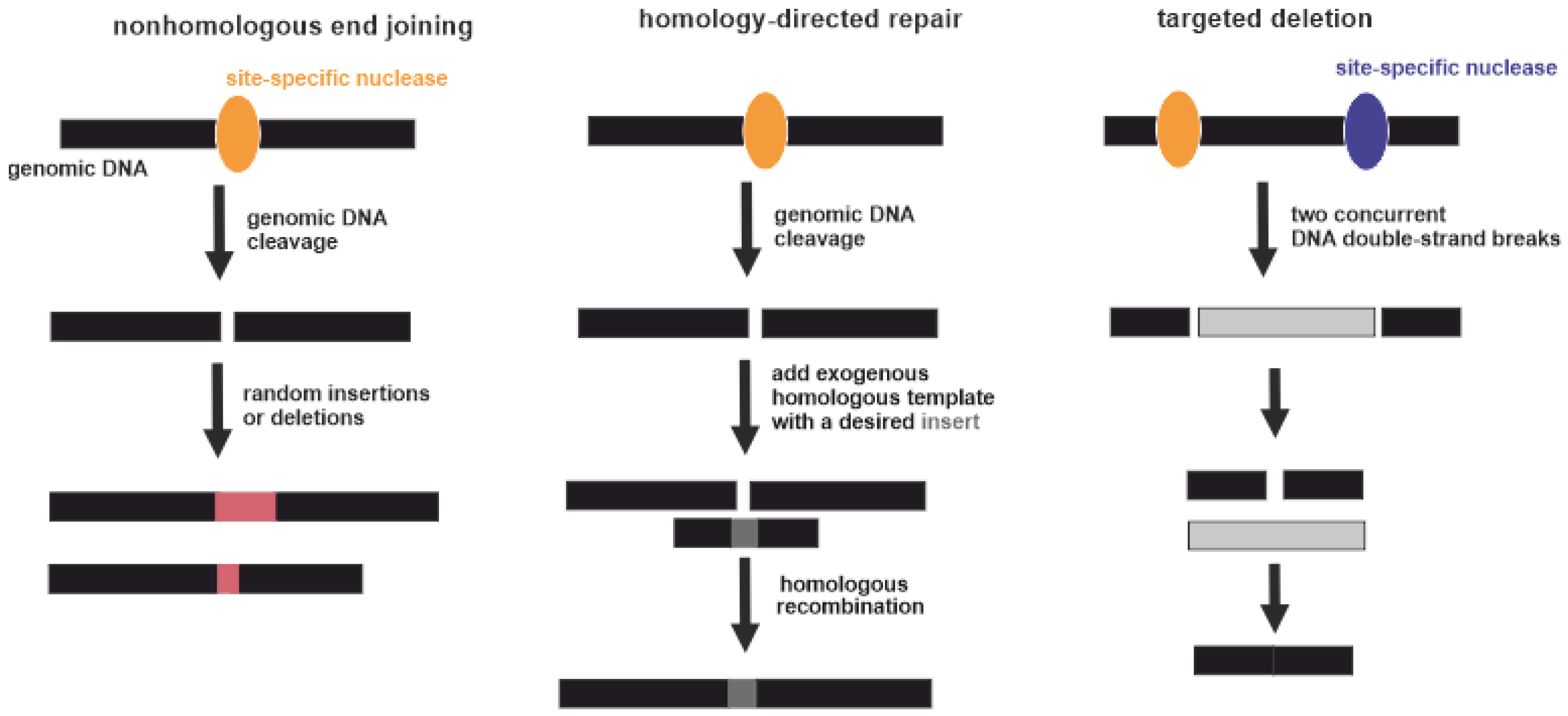
3.4. (Epi)Genome Editing
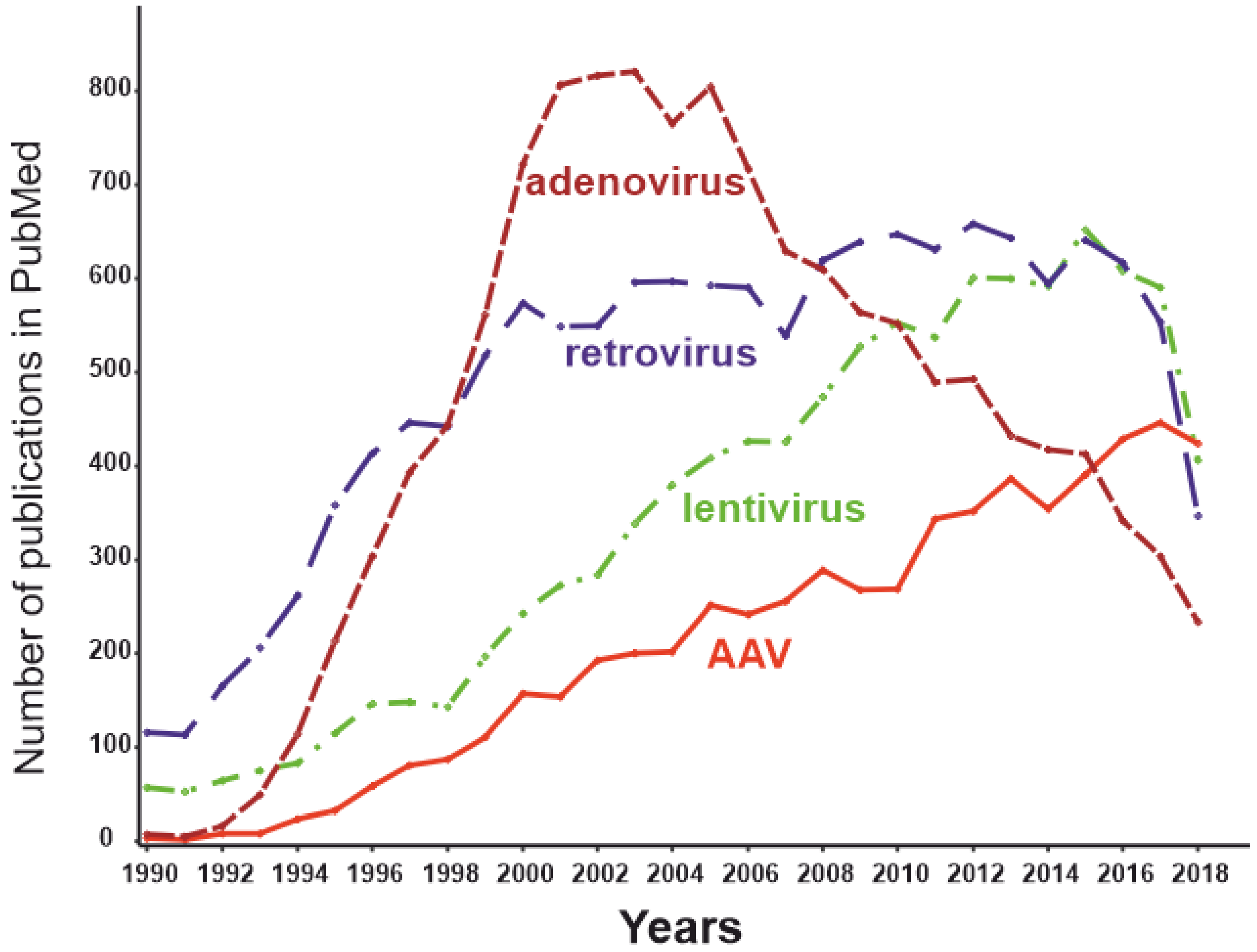
3.5. Viral Vectors
4. Future Directions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rosenberg, S.A.; Aebersold, P.; Cornetta, K.; Kasid, A.; Morgan, R.A.; Moen, R.; Karson, E.M.; Lotze, M.T.; Yang, J.C.; Topalian, S.L.; et al. Gene Transfer into Humans—Immunotherapy of Patients with Advanced Melanoma, Using Tumor-Infiltrating Lymphocytes Modified by Retroviral Gene Transduction. N. Engl. J. Med. 1990, 323, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.; Smith, E.; Hoy, D.; Carmona, L.; Wolfe, F.; Vos, T.; Williams, B.; Gabriel, S.; Lassere, M.; Johns, N.; et al. The global burden of rheumatoid arthritis: Estimates from the Global Burden of Disease 2010 study. Ann. Rheum. Dis. 2014, 73, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.L.; Vos, T.; Lozano, R.; Naghavi, M.; Flaxman, A.D.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2197–2223. [Google Scholar] [CrossRef]
- Nemtsova, M.V.; Zaletaev, D.V.; Bure, I.V.; Mikhaylenko, D.S.; Kuznetsova, E.B.; Alekseeva, E.A.; Beloukhova, M.I.; Deviatkin, A.A.; Lukashev, A.N.; Zamyatnin, A.A. Epigenetic Changes in the Pathogenesis of Rheumatoid Arthritis. Front. Genet. 2019, 10, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferucci, E.D.; Templin, D.W.; Lanier, A.P. Rheumatoid arthritis in American Indians and Alaska natives: A review of the literature. Semin. Arthritis Rheum. 2005, 34, 662–667. [Google Scholar] [CrossRef]
- Van Vollenhoven, R.F. Sex differences in rheumatoid arthritis: More than meets the eye. BMC Med. 2009, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.L.; Wolfe, F.; Huizinga, T.W.J. Rheumatoid arthritis. Lancet (Lond. Engl.) 2010, 376, 1094–1108. [Google Scholar] [CrossRef]
- Rainsford, K.D. Profile and mechanisms of gastrointestinal and other side effects of nonsteroidal anti-inflammatory drugs (NSAIDs). Am. J. Med. 1999, 107, 27–35. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Prim. 2018, 4, 18001. [Google Scholar] [CrossRef]
- Favalli, E.G.; Raimondo, M.G.; Becciolini, A.; Crotti, C.; Biggioggero, M.; Caporali, R. The management of first-line biologic therapy failures in rheumatoid arthritis: Current practice and future perspectives. Autoimmun. Rev. 2017, 16, 1185–1195. [Google Scholar] [CrossRef]
- KEGG. Available online: https://www.genome.jp/kegg-bin/show_pathway?hsa05323 (accessed on 14 October 2019).
- Aaltonen, K.J.; Ylikylä, S.; Tuulikki Joensuu, J.; Isomäki, P.; Pirilä, L.; Kauppi, M.; Rannio, T.; Eklund, K.; Blom, M.; Nordström, D. Efficacy and effectiveness of tumour necrosis factor inhibitors in the treatment of rheumatoid arthritis in randomized controlled trials and routine clinical practice. Rheumatology 2017, 56, 725–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
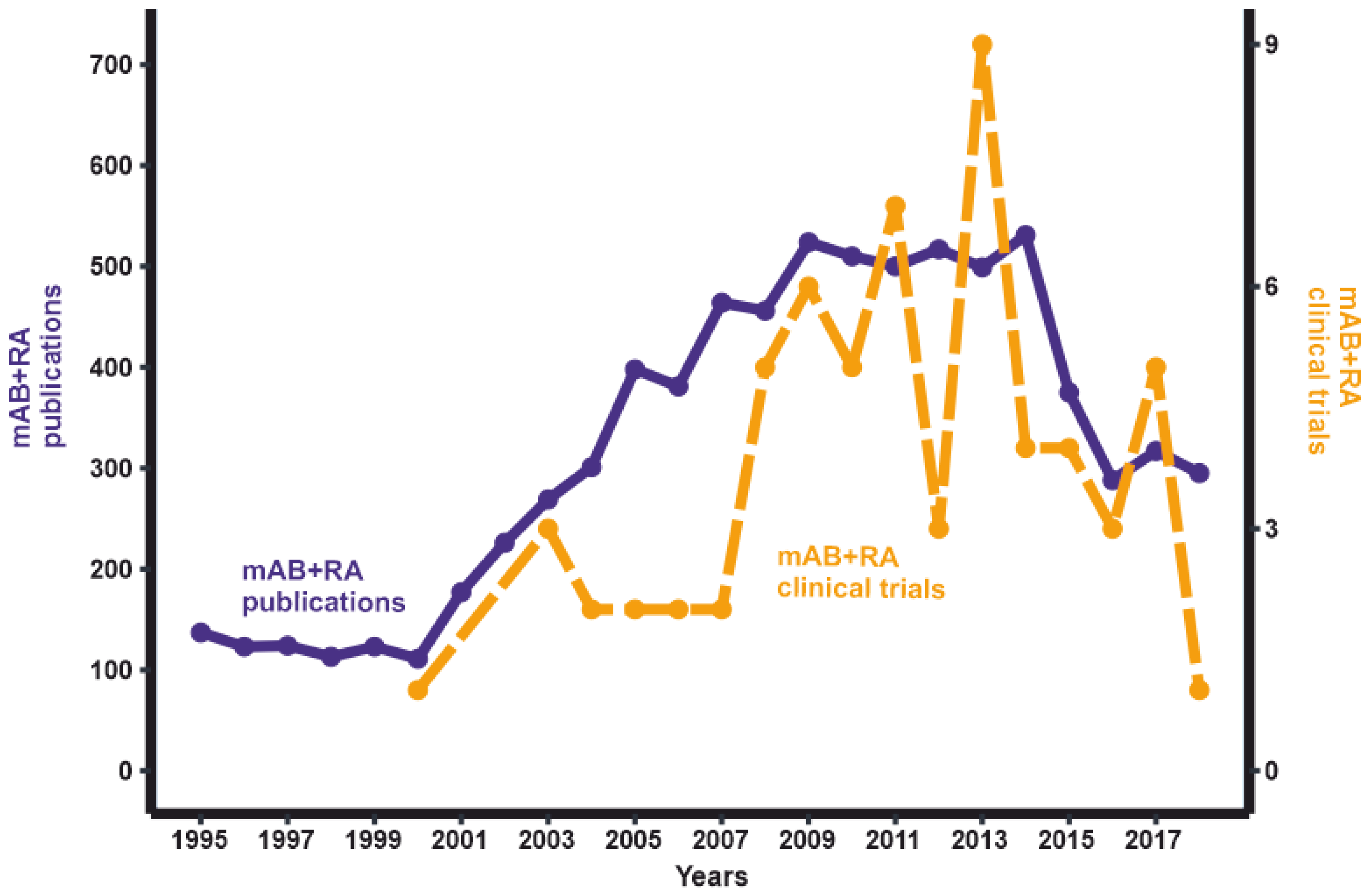
- ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/results?term=monoclonal+antibody&cond=Rheumatoid+Arthritis&Search=Apply&recrs=e&age_v=&gndr=&type=&rslt%20= (accessed on 14 October 2019).
- O’Shea, J.J.; Kontzias, A.; Yamaoka, K.; Tanaka, Y.; Laurence, A. Janus kinase inhibitors in autoimmune diseases. Ann. Rheum. Dis. 2013, 72 (Suppl. S2), ii111–ii115. [Google Scholar] [CrossRef] [PubMed]
- Bechman, K.; Yates, M.; Galloway, J.B. The new entries in the therapeutic armamentarium: The small molecule JAK inhibitors. Pharmacol. Res. 2019, 147, 104392. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K. Janus kinase inhibitors for rheumatoid arthritis. Curr. Opin. Chem. Biol. 2016, 32, 29–33. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef]
- Abbasi, M.; Mousavi, M.J.; Jamalzehi, S.; Alimohammadi, R.; Bezvan, M.H.; Mohammadi, H.; Aslani, S. Strategies toward rheumatoid arthritis therapy; the old and the new. J. Cell. Physiol. 2019, 234, 10018–10031. [Google Scholar] [CrossRef]
- Olsen, I.C.; Lie, E.; Vasilescu, R.; Wallenstein, G.; Strengholt, S.; Kvien, T.K. Assessments of the unmet need in the management of patients with rheumatoid arthritis: Analyses from the NOR-DMARD registry. Rheumatology 2019, 58, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Smeets, R.L.; van de Loo, F.A.J.; Arntz, O.J.; Bennink, M.B.; Joosten, L.A.B.; van den Berg, W.B. Adenoviral delivery of IL-18 binding protein C ameliorates Collagen-Induced Arthritis in mice. Gene Ther. 2003, 10, 1004–1011. [Google Scholar] [CrossRef]
- Plater-Zyberk, C.; Joosten, L.A.B.; Helsen, M.M.A.; Sattonnet-Roche, P.; Siegfried, C.; Alouani, S.; Van De Loo, F.A.J.; Graber, P.; Aloni, S.; Cirillo, R.; et al. Therapeutic effect of neutralizing endogenous IL-18 activity in the collagen-induced model of arthritis. J. Clin. Investig. 2001, 108, 1825–1832. [Google Scholar] [CrossRef]
- Palmer, G.; Talabot-Ayer, D.; Lamacchia, C.; Toy, D.; Seemayer, C.A.; Viatte, S.; Finckh, A.; Smith, D.E.; Gabay, C. Inhibition of interleukin-33 signaling attenuates the severity of experimental arthritis. Arthritis Rheum. 2009, 60, 738–749. [Google Scholar] [CrossRef]
- Hsu, Y.-H.; Hsieh, P.-P.; Chang, M.-S. Interleukin-19 blockade attenuates collagen-induced arthritis in rats. Rheumatology 2012, 51, 434–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00433875 (accessed on 21 October 2019).
- ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01636817 (accessed on 21 October 2019).
- ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00883896 (accessed on 21 October 2019).
- Blanco, F.J.; Möricke, R.; Dokoupilova, E.; Codding, C.; Neal, J.; Andersson, M.; Rohrer, S.; Richards, H. Secukinumab in Active Rheumatoid Arthritis: A Phase III Randomized, Double-Blind, Active Comparator– and Placebo-Controlled Study. Arthritis Rheumatol. 2017, 69, 1144–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, D.L.; Nguyen, K.H.Y.; Zhuang, S.; Shi, Y.; Mccormack, J.E.; Chada, S.; Firestein, G.S. Intra-articular IL-4 gene therapy in arthritis: Anti-inflammatory effect and enhanced Th2 activity. Gene Ther. 1999, 6, 1911–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Roon, J.A.; van Roy, J.L.; Gmelig-Meyling, F.H.; Lafeber, F.P.; Bijlsma, J.W. Prevention and reversal of cartilage degradation in rheumatoid arthritis by interleukin-10 and interleukin-4. Arthritis Rheum. 1996, 39, 829–835. [Google Scholar] [CrossRef]
- Steen-Louws, C.; Hartgring, S.A.Y.; Popov-Celeketic, J.; Lopes, A.P.; de Smet, M.B.M.; Eijkelkamp, N.; Lafeber, F.P.J.G.; Hack, C.E.; van Roon, J.A.G. IL4-10 fusion protein: A novel immunoregulatory drug combining activities of interleukin 4 and interleukin 10. Clin. Exp. Immunol. 2019, 195, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiolat, A.; Denys, A.; Petit, M.; Biton, J.; Lemeiter, D.; Herve, R.; Lutomski, D.; Boissier, M.-C.; Bessis, N. Interleukin-35 gene therapy exacerbates experimental rheumatoid arthritis in mice. Cytokine 2014, 69, 87–93. [Google Scholar] [CrossRef]
- Murphy, G.; Nagase, H. Reappraising metalloproteinases in rheumatoid arthritis and osteoarthritis: Destruction or repair? Nat. Clin. Pract. Rheumatol. 2008, 4, 128–135. [Google Scholar]
- Chubinskaya, S.; Kuettner, K.; Cole, A. Expression of matrix metalloproteinases in normal and damaged articular cartilage from human knee and ankle joints. Lab. Investig. 1999, 79, 1669–1677. [Google Scholar]
- Tchetverikov, I. Matrix metalloproteinases-3, -8, -9 as markers of disease activity and joint damage progression in early rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, 1094–1099. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Bay-Jensen, A.-C.; Karsdal, M.A.; Siebuhr, A.S.; Zheng, Q.; Maksymowych, W.P.; Christiansen, T.G.; Henriksen, K. The active form of MMP-3 is a marker of synovial inflammation and cartilage turnover in inflammatory joint diseases. BMC Musculoskelet. Disord. 2014, 15, 93. [Google Scholar] [CrossRef] [Green Version]
- Green, M.J. Serum MMP-3 and MMP-1 and progression of joint damage in early rheumatoid arthritis. Rheumatology 2003, 42, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, J.; Cawston, T. Matrix Metalloproteinase Knockout Studies and the Potential Use of Matrix Metalloproteinase Inhibitors in the Rheumatic Diseases. Curr. Drug Target Inflamm. Allergy 2005, 4, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. New strategies for targeting matrix metalloproteinases. Matrix Biol. 2015, 44–46, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; McKelvey, K.; Shen, K.; Minhas, N.; March, L.; Park, S.-Y.; Jackson, C.J. Endogenous MMP-9 and not MMP-2 promotes rheumatoid synovial fibroblast survival, inflammation and cartilage degradation. Rheumatology 2014, 53, 2270–2279. [Google Scholar] [CrossRef] [Green Version]
- Gossage, D.L.; Cieslarová, B.; Ap, S.; Zheng, H.; Xin, Y.; Lal, P.; Chen, G.; Smith, V.; Sundy, J.S. Phase 1b Study of the Safety, Pharmacokinetics, and Disease-related Outcomes of the Matrix Metalloproteinase-9 Inhibitor Andecaliximab in Patients With Rheumatoid Arthritis. Clin. Ther. 2018, 40, 156–165.e5. [Google Scholar] [CrossRef]
- Kaneko, K.; Williams, R.O.; Dransfield, D.T.; Nixon, A.E.; Sandison, A.; Itoh, Y. Selective Inhibition of Membrane Type 1 Matrix Metalloproteinase Abrogates Progression of Experimental Inflammatory Arthritis: Synergy With Tumor Necrosis Factor Blockade. Arthritis Rheumatol. 2016, 68, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, I.; Starskaia, I.; Nagy, T.; Guo, J.; Yatkin, E.; Väänänen, K.; Watford, W.T.; Chen, Z. Estrogen receptor contributes to T cell–mediated autoimmune inflammation by promoting T cell activation and proliferation. Sci. Signal. 2018, 11, eaap9415. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Sawada, T.; Nishiyama, S.; Shimada, K.; Tahara, K.; Hayashi, H.; Kato, E.; Tago, M.; Matsui, T.; Tohma, S. Influence of seasonal changes on disease activity and distribution of affected joints in rheumatoid arthritis. BMC Musculoskelet. Disord. 2019, 20, 30. [Google Scholar] [CrossRef]
- Watad, A.; Azrielant, S.; Bragazzi, N.L.; Sharif, K.; David, P.; Katz, I.; Aljadeff, G.; Quaresma, M.; Tanay, G.; Adawi, M.; et al. Seasonality and autoimmune diseases: The contribution of the four seasons to the mosaic of autoimmunity. J. Autoimmun. 2017, 82, 13–30. [Google Scholar] [CrossRef]
- Vieira, V.M.; Hart, J.E.; Webster, T.F.; Weinberg, J.; Puett, R.; Laden, F.; Costenbader, K.H.; Karlson, E.W. Association between residences in U.S. northern latitudes and rheumatoid arthritis: A spatial analysis of the nurses’ health study. Environ. Health Perspect. 2010, 118, 957–961. [Google Scholar] [CrossRef] [Green Version]
- Jahanban-Esfahlan, R.; Mehrzadi, S.; Reiter, R.J.; Seidi, K.; Majidinia, M.; Baghi, H.B.; Khatami, N.; Yousefi, B.; Sadeghpour, A. Melatonin in regulation of inflammatory pathways in rheumatoid arthritis and osteoarthritis: Involvement of circadian clock genes. Br. J. Pharmacol. 2018, 175, 3230–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Río, B.; Garćia Pedrero, J.M.; Martínez-Campa, C.; Zuazua, P.; Lazo, P.S.; Ramos, S. Melatonin, an endogenous-specific inhibitor of estrogen receptor α via calmodulin. J. Biol. Chem. 2004, 279, 38294–38302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, R.M.; Rao, D.S.; Baltimore, D. microRNA Regulation of Inflammatory Responses. Ann. Rev. Immunol. 2012, 30, 295–312. [Google Scholar] [CrossRef]
- Ceribelli, A.; Nahid, M.A.; Satoh, M.; Chan, E.K.L. MicroRNAs in rheumatoid arthritis. FEBS Lett. 2011, 585, 3667–3674. [Google Scholar] [CrossRef] [Green Version]
- Castro-Villegas, C.; Pérez-Sánchez, C.; Escudero, A.; Filipescu, I.; Verdu, M.; Ruiz-Limón, P.; Aguirre, M.A.; Jiménez-Gomez, Y.; Font, P.; Rodriguez-Ariza, A.; et al. Circulating miRNAs as potential biomarkers of therapy effectiveness in rheumatoid arthritis patients treated with anti-TNFα. Arthritis Res. Ther. 2015, 17, 49. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, N.; Kawakami, A. Recent findings regarding the effects of microRNAs on fibroblast-like synovial cells in rheumatoid arthritis. Immunol. Med. 2019, 42, 156–161. [Google Scholar] [CrossRef]
- Huizinga, T.; Nigrovic, P.; Ruderman, E.; Schulze-Koops, H. Altered expression of microRNA-203 in rheumatoid arthritis synovial fibroblasts and its role in fibroblast activation: Commentary. Int. J. Adv. Rheumatol. 2011, 9, 71–72. [Google Scholar]
- Trenkmann, M.; Brock, M.; Gay, R.E.; Michel, B.A.; Gay, S.; Huber, L.C. Tumor Necrosis Factor α-Induced MicroRNA-18a Activates Rheumatoid Arthritis Synovial Fibroblasts Through a Feedback Loop in NF-κB Signaling. Arthritis Rheum. 2013, 65, 916–927. [Google Scholar] [CrossRef]
- Akhtar, N.; Singh, A.K.; Ahmed, S. MicroRNA-17 Suppresses TNF-α Signaling by Interfering with TRAF2 and cIAP2 Association in Rheumatoid Arthritis Synovial Fibroblasts. J. Immunol. 2016, 197, 2219–2228. [Google Scholar] [CrossRef]
- Philippe, L.; Alsaleh, G.; Suffert, G.; Meyer, A.; Georgel, P.; Sibilia, J.; Wachsmann, D.; Pfeffer, S. TLR2 Expression Is Regulated by MicroRNA miR-19 in Rheumatoid Fibroblast-like Synoviocytes. J. Immunol. 2012, 188, 454–461. [Google Scholar] [CrossRef] [Green Version]
- Philippe, L.; Alsaleh, G.; Pichot, A.; Ostermann, E.; Zuber, G.; Frisch, B.; Sibilia, J.; Pfeffer, S.; Bahram, S.; Wachsmann, D.; et al. MiR-20a regulates ASK1 expression and TLR4-dependent cytokine release in rheumatoid fibroblast-like synoviocytes. Ann. Rheum. Dis. 2013, 72, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhang, Y.; Wu, J.; Jie, L.; Deng, J.; Zhao, D.; Yu, Q. Downregulated microRNA-135a ameliorates rheumatoid arthritis by inactivation of the phosphatidylinositol 3-kinase/AKT signaling pathway via phosphatidylinositol 3-kinase regulatory subunit 2. J. Cell. Physiol. 2019, 234, 17663–17676. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xian, P.-F.; Yang, L.; Wang, S.-X. MicroRNA-21 Promotes Proliferation of Fibroblast-Like Synoviocytes through Mediation of NF- κ B Nuclear Translocation in a Rat Model of Collagen-Induced Rheumatoid Arthritis. BioMed Res. Int. 2016, 2016, 9279078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Song, Q.; Shao, L.; Zhang, L.L.; Guo, X.H.; Mao, Y.J. MiR-124A inhibits proliferation and invasion of rheumatoid arthritis synovial fibroblasts. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4581–4588. [Google Scholar] [PubMed]
- Shi, D.L.; Shi, G.R.; Xie, J.; Du, X.Z.; Yang, H. MicroRNA-27a inhibits cell migration and invasion of fibroblast-like synoviocytes by targeting follistatin-like protein 1 in rheumatoid arthritis. Mol. Cells 2016, 39, 611–618. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cao, X. Long noncoding RNAs in innate immunity. Cell. Mol. Immunol. 2016, 13, 138–147. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Caffrey, D.R. Long noncoding RNAs in innate and adaptive immunity. Curr. Opin. Immunol. 2014, 26, 140–146. [Google Scholar] [CrossRef] [Green Version]
- Hur, K.; Kim, S.H.; Kim, J.M. Potential implications of long noncoding rnas in autoimmune diseases. Immune Netw. 2019, 19, e4. [Google Scholar] [CrossRef]
- Wu, G.-C.; Pan, H.-F.; Leng, R.-X.; Wang, D.-G.; Li, X.-P.; Li, X.-M.; Ye, D.-Q. Emerging role of long noncoding RNAs in autoimmune diseases. Autoimmun. Rev. 2015, 14, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.; Döring, F.; Klapper, M.; Neumann, K.; Schulte, D.M.; Türk, K.; Schröder, J.O.; Zeuner, R.A.; Freitag-Wolf, S.; Schreiber, S.; et al. Interleukin-6 and Tumour Necrosis Factor-α differentially regulate lincRNA transcripts in cells of the innate immune system in vivo in human subjects with rheumatoid arthritis. Cytokine 2014, 68, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kim, D.; Han, J.; Kim, Y.; Lee, M.; Jin, E.-J. PBMC and exosome-derived Hotair is a critical regulator and potent marker for rheumatoid arthritis. Clin. Exp. Med. 2015, 15, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; He, X.; Chen, Z.; Fan, D.; Wang, Y.; Feng, H.; Zhang, G.; Lu, A.; Xiao, L. LncRNA HOTAIR-mediated Wnt/β-catenin network modeling to predict and validate therapeutic targets for cartilage damage. BMC Bioinform. 2019, 20, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, B.Y.; Guo, X.H.; Yang, M.R.; Liu, F.; Bi, X.; Liu, Y.; Fang, L.K.; Luo, X.Q.; Wang, J.; Bellanti, J.A.; et al. Long Non-Coding RNA GAPLINC Promotes Tumor-Like Biologic Behaviors of Fibroblast-Like Synoviocytes as MicroRNA Sponging in Rheumatoid Arthritis Patients. Front. Immunol. 2018, 9, 702. [Google Scholar] [CrossRef]
- Zou, Y.; Xu, S.; Xiao, Y.; Qiu, Q.; Shi, M.; Wang, J.; Liang, L.; Zhan, Z.; Yang, X.; Olsen, N.; et al. Long noncoding RNA LERFS negatively regulates rheumatoid synovial aggression and proliferation. J. Clin. Investig. 2018, 128, 4510–4524. [Google Scholar] [CrossRef] [Green Version]
- Shui, X.; Chen, S.; Lin, J.; Kong, J.; Zhou, C.; Wu, J. Knockdown of lncRNA NEAT1 inhibits Th17/CD4 + T cell differentiation through reducing the STAT3 protein level. J. Cell. Physiol. 2019, 234, 22477–22484. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.Z.; Sun, N.; Liu, J.H.; Chen, F.F.; Guan, X.L.; Li, A.; Wang, F.; Zhao, Q.F.; Wang, H.Y.; et al. Long noncoding RNA expression profile in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 227. [Google Scholar] [CrossRef] [Green Version]
- Quek, X.C.; Thomson, D.W.; Maag, J.L.V.; Bartonicek, N.; Signal, B.; Clark, M.B.; Gloss, B.S.; Dinger, M.E. lncRNAdb v2.0: Expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 2015, 43, D168–D173. [Google Scholar] [CrossRef]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef]
- Danan, M.; Schwartz, S.; Edelheit, S.; Sorek, R. Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res. 2012, 40, 3131–3142. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Holdt, L.M.; Kohlmaier, A.; Teupser, D. Circular RNAs as therapeutic agents and targets. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Starke, S.; Jost, I.; Rossbach, O.; Schneider, T.; Schreiner, S.; Hung, L.-H.; Bindereif, A. Exon Circularization Requires Canonical Splice Signals. Cell Rep. 2015, 10, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Barrett, S.P.; Wang, P.L.; Salzman, J. Circular RNA biogenesis can proceed through an exon-containing lariat precursor. Elife 2015, 4, e07540. [Google Scholar] [CrossRef]
- Schindewolf, C.; Braun, S.; Domdey, H. In vitro Generation of a Circular Exon from a Linear Pre-mRNA Transcript. Nucleic Acids Res. 1996, 24, 1260–1266. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Tang, X.; Wang, S. Roles of CircRNAs in autoimmune diseases. Front. Immunol. 2019, 10, 639. [Google Scholar] [CrossRef]
- Zhou, Z.; Sun, B.; Huang, S.; Zhao, L. Roles of circular RNAs in immune regulation and autoimmune diseases. Cell Death Dis. 2019, 10, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, F.; Yu, X.; Huang, J.; Dai, Y. Circular RNA expression profiles of peripheral blood mononuclear cells in rheumatoid arthritis patients, based on microarray chip technology. Mol. Med. Rep. 2017, 16, 8029–8036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-C.; Lee, Y.-C.; Chang, K.-L.; Hsiao, K.-Y. Analysis of common targets for circular RNAs. BMC Bioinform. 2019, 20, 372. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Fan, Y.; Liu, Y.; Xie, W.; Zhang, Z. Efficacy and safety of secukinumab in active rheumatoid arthritis with an inadequate response to tumor necrosis factor inhibitors: A meta-analysis of phase III randomized controlled trials. Clin. Rheumatol. 2019, 38, 2765–2776. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.F.; Zhao, X.Y.; Liu, W.; Liu, X.P. UCA1 impacts progress of rheumatoid arthritis by inducing the apoptosis of fibroblast-like synoviocyte. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 914–920. [Google Scholar] [PubMed]
- Ye, Y.; Gao, X.; Yang, N. LncRNA ZFAS1 promotes cell migration and invasion of fibroblast-like synoviocytes by suppression of miR-27a in rheumatoid arthritis. Hum. Cell 2018, 31, 14–21. [Google Scholar] [CrossRef]
- Collins, M.; Thrasher, A. Gene therapy: Progress and predictions. Proc. R. Soc. B Biol. Sci. 2015, 282, 20143003. [Google Scholar] [CrossRef] [Green Version]
- Zsebo, K.; Yaroshinsky, A.; Rudy, J.J.; Wagner, K.; Greenberg, B.; Jessup, M.; Hajjar, R.J. Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: Analysis of recurrent cardiovascular events and mortality. Circ. Res. 2014, 114, 101–108. [Google Scholar] [CrossRef]
- Rodrigues, G.A.; Shalaev, E.; Karami, T.K.; Cunningham, J.; Slater, N.K.H.; Rivers, H.M. Pharmaceutical Development of AAV-Based Gene Therapy Products for the Eye. Pharm. Res. 2019, 36, 29. [Google Scholar] [CrossRef] [Green Version]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.D.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef] [Green Version]
- Van Riel, P.L.C.M.; Renskers, L. The Disease Activity Score (DAS) and the Disease Activity Score using 28 joint counts (DAS28) in the management of rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34 (Suppl. S101), S40–S44. [Google Scholar]
- Fuchs, H.A.; Brooks, R.H.; Callahan, L.F.; Pincus, T. A simplified twenty-eight–joint quantitative articular index in rheumatoid arthritis. Arthritis Rheum. 1989, 32, 531–537. [Google Scholar] [CrossRef]
- Woods, J.; Sitabkhan, Y.; Koch, A. Gene Therapy for Rheumatoid Arthritis: Recent Advances. Curr. Gene Ther. 2008, 8, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Challis, R.C.; Ravindra Kumar, S.; Chan, K.Y.; Challis, C.; Beadle, K.; Jang, M.J.; Kim, H.M.; Rajendran, P.S.; Tompkins, J.D.; Shivkumar, K.; et al. Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat. Protoc. 2019, 14, 379–414. [Google Scholar] [CrossRef] [PubMed]
- Kaji, E.H.; Leiden, J.M. Gene and Stem Cell Therapies. JAMA 2001, 285, 545–550. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.H.; Ghivizzani, S.C.; Robbins, P.D. Gene Delivery to Joints by Intra-articular Injection. Hum. Gene Ther. 2017, 29, 2–14. [Google Scholar] [CrossRef]
- Rai, M.F.; Pan, H.; Yan, H.; Sandell, L.J.; Pham, C.T.N.; Wickline, S.A. Applications of RNA interference in the treatment of arthritis. Transl. Res. 2019, 214, 1–16. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Ann. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Keam, S.J. Inotersen: First Global Approval. Drugs 2018, 78, 1371–1376. [Google Scholar] [CrossRef]
- Banerjee, D.; Roeker, L.E.; Grogan, M.; Swiecicki, P.; Poterucha, J.; Heimbach, J.; Zeldenrust, S.; Gertz, M.; Edwards, B.; Daly, R.; et al. Outcomes of patients with familial transthyretin amyloidosis after liver transplantation. Prog. Transplant. 2017, 27, 246–250. [Google Scholar] [CrossRef]
- Gertz, M.A.; Mauermann, M.L.; Grogan, M.; Coelho, T. Advances in the treatment of hereditary transthyretin amyloidosis: A review. Brain Behav. 2019, 9, e01371. [Google Scholar] [CrossRef] [Green Version]
- Wesselhoeft, R.A.; Kowalski, P.S.; Anderson, D.G. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat. Commun. 2018, 9, 2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragomir, M.; Calin, G.A. Circular RNAs in cancer—Lessons learned from microRNAs. Front. Oncol. 2018, 8, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Bhan, A.K.; Deshpande, V.; Shankar, P.; Manjunath, N. Silencing TNF-α in macrophages and dendritic cells for arthritis treatment. Scand. J. Rheumatol. 2013, 42, 266–269. [Google Scholar] [CrossRef]
- Zhou, H.; Yan, H.; Pan, H.; Hou, K.K.; Akk, A.; Springer, L.E.; Hu, Y.; Allen, J.S.; Wickline, S.A.; Pham, C.T.N. Peptide-siRNA nanocomplexes targeting NF-κB subunit p65 suppress nascent experimental arthritis. J. Clin. Investig. 2014, 124, 4363–4374. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Lee, A.; Hwang, S.R.; Park, J.-S.; Jang, J.; Huh, M.S.; Jo, D.-G.; Yoon, S.-Y.; Byun, Y.; Kim, S.H.; et al. TNF-α Gene Silencing Using Polymerized siRNA/Thiolated Glycol Chitosan Nanoparticles for Rheumatoid Arthritis. Mol. Ther. 2014, 22, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Chen, Y.; Jiang, S.; Yang, K.; Li, X.; Zhao, X.; Ouyang, Y.; Fan, C.; Yuan, W. Efficient and Non-Toxic Biological Response Carrier Delivering TNF-α shRNA for Gene Silencing in a Murine Model of Rheumatoid Arthritis. Front. Immunol. 2016, 7, 305. [Google Scholar] [CrossRef] [Green Version]
- Setten, R.L.; Rossi, J.J.; Han, S. ping The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef] [Green Version]
- Adkar, S.S.; Brunger, J.M.; Willard, V.P.; Wu, C.L.; Gersbach, C.A.; Guilak, F. Genome Engineering for Personalized Arthritis Therapeutics. Trends Mol. Med. 2017, 23, 917–931. [Google Scholar] [CrossRef]
- Zhang, H.X.; Zhang, Y.; Yin, H. Genome Editing with mRNA Encoding ZFN, TALEN, and Cas9. Mol. Ther. 2019, 27, 735–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Eyre, S.; Suzuki, A.; Kochi, Y.; Yamamoto, K. Genetics of rheumatoid arthritis: 2018 status. Ann. Rheum. Dis. 2019, 78, 446–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigt, P.; Reinberg, D. Epigenome editing. Nat. Biotechnol. 2013, 31, 1097–1099. [Google Scholar] [CrossRef]
- Knowles, M.R.; Hohneker, K.W.; Zhou, Z.; Olsen, J.C.; Noah, T.L.; Hu, P.-C.; Leigh, M.W.; Engelhardt, J.F.; Edwards, L.J.; Jones, K.R.; et al. A Controlled Study of Adenoviral-Vector–Mediated Gene Transfer in the Nasal Epithelium of Patients with Cystic Fibrosis. N. Engl. J. Med. 1995, 333, 823–831. [Google Scholar] [CrossRef]
- Morgan, J.E. Cell and Gene Therapy in Duchenne Muscular Dystrophy. Hum. Gene Ther. 1994, 5, 165–173. [Google Scholar] [CrossRef]
- Grossman, M.; Raper, S.E.; Kozarsky, K.; Stein, E.A.; Engelhardt, J.F.; Muller, D.; Lupien, P.J.; Wilson, J.M. Successful ex vivo gene therapy directed to liver in a patient with familial hypercholesterolaemia. Nat. Genet. 1994, 6, 335–341. [Google Scholar] [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [Green Version]
- Ni, R.; Zhou, J.; Hossain, N.; Chau, Y. Virus-inspired nucleic acid delivery system: Linking virus and viral mimicry. Adv. Drug Deliv. Rev. 2016, 106, 3–26. [Google Scholar] [CrossRef]
- Lukashev, A.N.; Zamyatnin, A.A. Viral vectors for gene therapy: Current state and clinical perspectives. Biochemistry 2016, 81, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Lehrman, S. Virus treatment questioned after gene therapy death. Nature 1999, 401, 517–518. [Google Scholar] [CrossRef] [PubMed]
- Quantin, B.; Perricaudet, L.D.; Tajbakhsh, S.; Mandel, J.L. Adenovirus as an expression vector in muscle cells in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 2581–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.D.; Ghivizzani, S.C. Viral vectors for gene therapy. Pharmacol. Ther. 1998, 80, 35–47. [Google Scholar] [CrossRef]
- Blaese, R.M.; Culver, K.W.; Miller, A.D.; Carter, C.S.; Fleisher, T.; Clerici, M.; Shearer, G.; Chang, L.; Chiang, Y.; Tolstoshev, P.; et al. T Lymphocyte-Directed Gene Therapy for ADA- SCID: Initial Trial Results After 4 Years. Science 1995, 270, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.-L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A Serious Adverse Event after Successful Gene Therapy for X-Linked Severe Combined Immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef] [Green Version]
- Hacein-Bey-Abina, S.; Le Deist, F.; Carlier, F.; Bouneaud, C.; Hue, C.; De Villartay, J.-P.; Thrasher, A.J.; Wulffraat, N.; Sorensen, R.; Dupuis-Girod, S.; et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N. Engl. J. Med. 2002, 346, 1185–1193. [Google Scholar] [CrossRef]
- Iwakuma, T.; Cui, Y.; Chang, L.-J. Self-Inactivating Lentiviral Vectors with U3 and U5 Modifications. Virology 1999, 261, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Wylie, K.M.; Mihindukulasuriya, K.A.; Zhou, Y.; Sodergren, E.; Storch, G.A.; Weinstock, G.M. Metagenomic analysis of double-stranded DNA viruses in healthy adults. BMC Biol. 2014, 12, 71. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Schaffer, D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014, 15, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Kaplitt, M.G.; Leone, P.; Samulski, R.J.; Xiao, X.; Pfaff, D.W.; O’Malley, K.L.; During, M.J. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat. Genet. 1994, 8, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, S.; Mizukami, H.; Ogura, T.; Kure, S.; Ichinohe, A.; Kojima, K.; Matsubara, Y.; Kobayahi, E.; Okada, T.; Hoshika, A.; et al. Long-term correction of hyperphenylalaninemia by AAV-mediated gene transfer leads to behavioral recovery in phenylketonuria mice. Gene Ther. 2004, 11, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, J.; Hong, W.; Zhang, P.; Wang, X.; Körner, H.; Wei, W. Ontology and Function of Fibroblast-Like and Macrophage-Like Synoviocytes: How Do They Talk to Each Other and Can They Be Targeted for Rheumatoid Arthritis Therapy? Front. Immunol. 2018, 9, 1467. [Google Scholar] [CrossRef]
- Jubb, Kennedy & Palmer’s Pathology of Domestic Animals: Volume 2, 6th ed.; Maxie, M.G. (Ed.) Saunders Ltd.: Maryland Heights, MO, USA, 2016; ISBN 9780702053184. [Google Scholar]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, H.; Colosi, P. Effect of Genome Size on AAV Vector Packaging. Mol. Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef] [Green Version]
- Potter, R.A.; Griffin, D.A.; Sondergaard, P.C.; Johnson, R.W.; Pozsgai, E.R.; Heller, K.N.; Peterson, E.L.; Lehtimäki, K.K.; Windish, H.P.; Mittal, P.J.; et al. Systemic Delivery of Dysferlin Overlap Vectors Provides Long-Term Gene Expression and Functional Improvement for Dysferlinopathy. Hum. Gene Ther. 2017, 29, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Drouin, L.M.; Agbandje-McKenna, M. Adeno-associated virus structural biology as a tool in vector development. Future Virol. 2013, 8, 1183–1199. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated Virus Serotypes: Vector Toolkit for Human Gene Therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Deverman, B.E.; Pravdo, P.L.; Simpson, B.P.; Kumar, S.R.; Chan, K.Y.; Banerjee, A.; Wu, W.L.; Yang, B.; Huber, N.; Pasca, S.P.; et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016, 34, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Hemphill, D.D.; McIlwraith, C.W.; Samulski, R.J.; Goodrich, L.R. Adeno-associated viral vectors show serotype specific transduction of equine joint tissue explants and cultured monolayers. Sci. Rep. 2014, 4, 5861. [Google Scholar] [CrossRef] [PubMed]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of Serum IgG and Neutralizing Factors Against Adeno-Associated Virus (AAV) Types 1, 2, 5, 6, 8, and 9 in the Healthy Population: Implications for Gene Therapy Using AAV Vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Hüser, D.; Khalid, D.; Lutter, T.; Hammer, E.-M.; Weger, S.; Heßler, M.; Kalus, U.; Tauchmann, Y.; Hensel-Wiegel, K.; Lassner, D.; et al. High Prevalence of Infectious Adeno-associated Virus (AAV) in Human Peripheral Blood Mononuclear Cells Indicative of T Lymphocytes as Sites of AAV Persistence. J. Virol. 2017, 91, 302–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchlis, G.; Podsakoff, G.M.; Radu, A.; Hawk, S.M.; Flake, A.W.; Mingozzi, F.; High, K.A. Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood 2012, 119, 3038–3041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivière, C.; Danos, O.; Douar, A.M. Long-term expression and repeated administration of AAV type 1, 2 and 5 vectors in skeletal muscle of immunocompetent adult mice. Gene Ther. 2006, 13, 1300–1308. [Google Scholar] [CrossRef] [Green Version]
- Tse, L.V.; Klinc, K.A.; Madigan, V.J.; Rivera, R.M.C.; Wells, L.F.; Havlik, L.P.; Smith, J.K.; Agbandje-McKenna, M.; Asokan, A. Structure-guided evolution of antigenically distinct adeno-associated virus variants for immune evasion. Proc. Natl. Acad. Sci. USA 2017, 114, E4812–E4821. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, J.; Liu, Y.; Shi, Z.; Liu, H.; Wei, Y.; Yang, L. Bat adeno-associated viruses as gene therapy vectors with the potential to evade human neutralizing antibodies. Gene Ther. 2019, 26, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Barnes, C.; Scheideler, O.; Schaffer, D. Engineering the AAV capsid to evade immune responses. Curr. Opin. Biotechnol. 2019, 60, 99–103. [Google Scholar] [CrossRef]
- Meliani, A.; Boisgerault, F.; Hardet, R.; Marmier, S.; Collaud, F.; Ronzitti, G.; Leborgne, C.; Costa Verdera, H.; Simon Sola, M.; Charles, S.; et al. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat. Commun. 2018, 9, 4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundstrom, K. RNA viruses as tools in gene therapy and vaccine development. Genes 2019, 10, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wold, W.S.M.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Subramanian, G.; Silayeva, L.; Newkirk, I.; Doctor, D.; Chawla, K.; Chattopadhyay, S.; Chandra, D.; Chilukuri, N.; Betapudi, V. Gene therapy leaves a vicious cycle. Front. Oncol. 2019, 9, 297. [Google Scholar] [CrossRef]
- Fan, H.; Zhao, Z.; Yan, G.; Zhang, X.; Yang, C.; Meng, H.; Chen, Z.; Liu, H.; Tan, W. A Smart DNAzyme-MnO 2 Nanosystem for Efficient Gene Silencing. Angew. Chem. Int. Ed. 2015, 54, 4801–4805. [Google Scholar] [CrossRef]
- Fan, H.; Zhang, X.; Lu, Y. Recent advances in DNAzyme-based gene silencing. Sci. China Chem. 2017, 60, 591–601. [Google Scholar] [CrossRef]
- Karuppal, R. Current concepts in the articular cartilage repair and regeneration. J. Orthop. 2017, 14, A1–A3. [Google Scholar] [CrossRef]
- King, B.L.; Yin, V.P. A conserved microRNA regulatory circuit is differentially controlled during limb/appendage regeneration. PLoS ONE 2016, 11, e0157106. [Google Scholar] [CrossRef] [Green Version]
- Hsueh, M.-F.; Önnerfjord, P.; Bolognesi, M.P.; Easley, M.E.; Kraus, V.B. Analysis of “old” proteins unmasks dynamic gradient of cartilage turnover in human limbs. Sci. Adv. 2019, 5, eaax3203. [Google Scholar] [CrossRef] [Green Version]
- Imaeda, A.; Tomoike, F.; Hayakawa, M.; Nakamoto, K.; Kimura, Y.; Abe, N.; Abe, H. N6-methyl adenosine in siRNA evades immune response without reducing RNAi activity. Nucleosides Nucleotides Nucleic Acids 2019, 11, e0157106. [Google Scholar] [CrossRef]
- Vlachogiannis, N.I.; Gatsiou, A.; Silvestris, D.A.; Stamatelopoulos, K.; Tektonidou, M.G.; Gallo, A.; Sfikakis, P.P.; Stellos, K. Increased adenosine-to-inosine RNA editing in rheumatoid arthritis. J. Autoimmun. 2019, 106, 102329. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Molecule | Possible Target | Effector | Model | Therapeutic Effect | References |
|---|---|---|---|---|---|
| Cytokine | IL-17 | Human anti-IL-17 mAB (secukinumab) | Phase III clinical trials, patients with RA who have inadequate response to anti-TNF therapy | Significantly better clinical efficacy as compared with placebo, but the effect does not exceed that of treatment with abatacept. | [27,87] |
| Cytokine | IL-4, IL-10 | IL4-10 FP | Mice, PGIA | Suppression of disease severity without inducing B cell hyperactivity. Suppression of articular cartilage damage in models for osteoarthritis. | [30] |
| MMP | MT1-MMP | MT1-MMP selective inhibitory antibody (DX-2400) and/or TNFR-Fc fusion protein | Mice, CIA | Reduction of cartilage degradation and disease progression. DX-2400 and TNFR-Fc acted synergistically. | [41] |
| MMP | MMP-9 | mAB to MMP-9 (andecaliximab) | Phase Ib completed | Proved short-term safety; further studies are warranted. | [40] |
| miRNA | miR-203 | Transfection with synthetic miR-203 precursor | Human, primary FLSs culture (RA and HC) | Overexpression in RA-FLS led to increased levels of MMP1, IL-6. | [53] |
| miRNA | miR-18a | Transfection with synthetic miR-18a precursor | Human, primary RA-FLSs culture | Overexpression in RA-FLS led to increased levels of MMP1, IL-6, and IL-8. | [54] |
| miRNA | miR-17 | Transfection with synthetic miR-17 precursor | Human, primary RA-FLSs culture | Inhibited the TNF-α-induced IL-6, IL-8, MMP-1, and MMP-13 production in RA-FLSs. | [55] |
| miRNA | miR-19a/b | Transfection with miR-19a and miR-19b mimics | Human, primary RA-FLSs culture | Overexpression led to downregulation of IL-6 and MMP-3 secretion by controlling TLR2 expression. | [56] |
| miRNA | miR-20a | Transfection with miR-20 mimics | Human, primary RA-FLSs culture | Overexpression led to decreased IL-6 and CXCL10 release by RA-FLS. | [57] |
| miRNA | miR-135a | siRNA | Human, primary FLSs culture | Downregulation of miR-135a led to inhibited cell proliferation, migration, and invasion and promoted cell apoptosis through upregulation of PIK3R2 and inactivation of the PI3K/AKT signaling pathway. | [58] |
| miRNA | miR-21 | siRNA (lentivirus) | Rats, CIA | Inhibition of miR-21 in RA-FLSs led to significant decrease in cell proliferation rates. | [59] |
| miRNA | miR-124a | Chemically synthetized miR-124a mimic | Human, primary RA-FLSs culture | Suppresses the proliferation and invasion of RA-FLSs | [60] |
| miRNA | miR-27a | Transfection with miR-27a | Human, primary RA-FLSs culture | Overexpression inhibited cell migration and invasion of RA-FLSs by targeting FSTL1 and restraining the TLR-4/NF-κB pathway. | [61] |
| lncRNA | GAPLINC | siRNA | Human, primary RA-FLSs culture | Decreased the migration and invasion of RA-FLSs as well as production of proinflammatory cytokines (IL-6 and IL-8) and MMPs. | [70] |
| lncRNA | NEAT1 | Lentivirus-constructed short hairpin RNA interference, injection into joint | Mice, CIA | Inhibited differentiation of CD4+ T cells into Th17 cells through reducing level of STAT3 transcription factor. | [72] |
| lncRNA | UCA1 | siRNA | Human, primary RA-FLSs culture | The downregulation of UCA1 expression increased the viability in normal FLSs, while overexpression of UCA1 in RA-FLSs inhibited the viability of cells. | [88] |
| lncRNA | LERFS | lncRNA Smart Silencer (RiboBio) | Human, primary FLSs culture (RA and HC) | Silencing of LERFS led to increased proliferation and migration of FLSs. | [71] |
| lncRNA | ZFAS1 | shRNAs (lentivirus) | Human, primary FLSs culture (RA and HC) | Knockdown decreased MMP-2 and MMP-9 expression and thus suppressed migration and invasion of RA-FLSs through suppression of miR-27a. | [89] |
| Type of Viral Vector | Unique Properties | Side Effects | Clinical Implication for RA Gene Therapy |
|---|---|---|---|
| Adenovirus | 37-kb-large insert size for the gene of interest; adenoviruses are maintained in cells as an episome [159] | Potent immune response in the case of a systemically administered adenoviral vector | Adenoviral vectors are used primarily for applications in which an immune response is desirable. Autoimmune RA is not among the optimal applications for adenoviral delivery systems. |
| Retrovirus | 9–12-kb-large insert size for the gene of interest; viral DNA is integrated into the host genome [160] | High risk of malignancies | The risk of insertional mutagenesis for retroviral vectors limits their clinical implication. |
| Lentivirus | 9–12-kb-large insert size for the gene of interest; viral DNA is integrated into the host genome [160] | Risk of malignancies | Introduction of lentivirus-based constructs into clinical practice is limited and must include a very careful risk/benefit analysis, which is unlikely to be favorable in the case of RA. |
| AAV | 4.8-kb-large insert size for the gene of interest [124];AAV genome mostly exists as an epichromosome | Good safety profile | AAV vectors are recognized as safe and effective, being one of the most promising methods of gene therapy. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deviatkin, A.A.; Vakulenko, Y.A.; Akhmadishina, L.V.; Tarasov, V.V.; Beloukhova, M.I.; Zamyatnin Jr., A.A.; Lukashev, A.N. Emerging Concepts and Challenges in Rheumatoid Arthritis Gene Therapy. Biomedicines 2020, 8, 9. https://doi.org/10.3390/biomedicines8010009
Deviatkin AA, Vakulenko YA, Akhmadishina LV, Tarasov VV, Beloukhova MI, Zamyatnin Jr. AA, Lukashev AN. Emerging Concepts and Challenges in Rheumatoid Arthritis Gene Therapy. Biomedicines. 2020; 8(1):9. https://doi.org/10.3390/biomedicines8010009
Chicago/Turabian StyleDeviatkin, Andrei A., Yulia A. Vakulenko, Ludmila V. Akhmadishina, Vadim V. Tarasov, Marina I. Beloukhova, Andrey A. Zamyatnin Jr., and Alexander N. Lukashev. 2020. "Emerging Concepts and Challenges in Rheumatoid Arthritis Gene Therapy" Biomedicines 8, no. 1: 9. https://doi.org/10.3390/biomedicines8010009